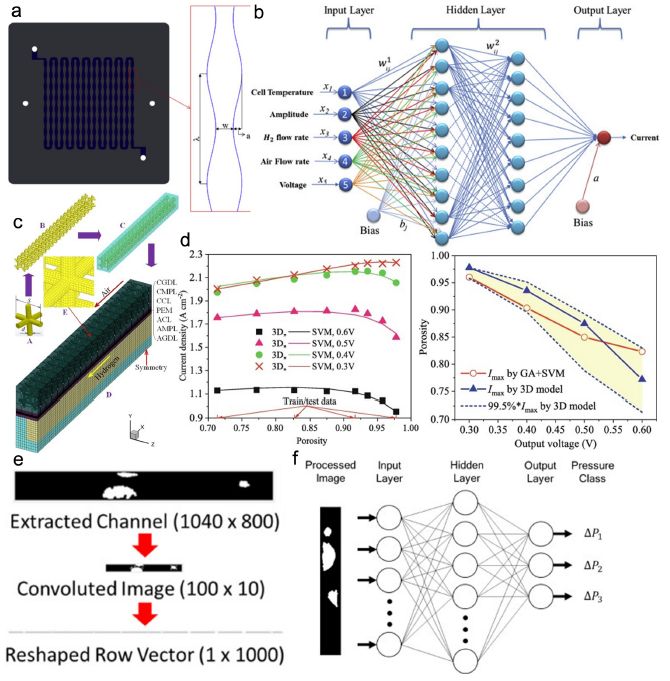
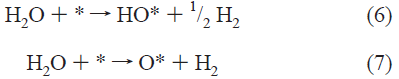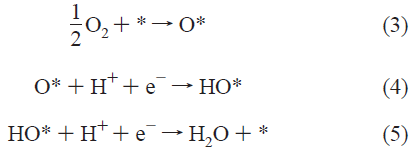燃料電池触媒の構造と特性の関係を知りたいと思っていくつか論文を読んできたが、いまだ何も理解できていない。
Resolving the nanoparticles’ structure-property relationships at the atomic level: a study of Pt-based electrocatalysts
この論文を読んでみよう。
SUMMARY
Achieving highly active and stable oxygen reduction reaction performance at low
platinum-group-metal loadings remains one of the grand challenges in the proton-exchange membrane fuel cells community.
Currently, state-of-the-art electrocatalysts are high-surface-area-carbon-supported nanoalloys of platinum with different transition metals (Cu, Ni, Fe, and Co).
Despite years of focused research, the established structure-property relationships are not able to explain and predict the electrochemical performance and behavior of the real nanoparticulate systems.
In the first part of this work, we reveal the complexity of commercially available platinum-based electrocatalysts and their electrochemical behavior.
In the second part, we introduce a bottom-up approach where atomically resolved properties, structural changes, and strain analysis are recorded as well as analyzed on an individual nanoparticle before and after electrochemical conditions (e.g. high current density).
Our methodology offers a new level of understanding of structure-stability relationships of practically viable nanoparticulate systems.
INTRODUCTION
In the race toward cutting the World’s greenhouse gas emissions, low-temperature proton-exchange membrane fuel cells (LT-PEMFCs) and batteries are needed to carry out electrochemical reactions to transform and store sustainably produced energy (Gro¨ ger et al., 2015).
Although PEMFC technology is finally on the cusp of mass adoption, its high cost is becoming a major bottleneck—with Pt-based electrocatalyst contributing almost
50% of the total cost of PEMFC system manufacturing (DOE, 2017).
The majority of these Pt-based electrocatalysts are necessary to catalyze the sluggish cathodic oxygen reduction reaction (ORR).
This motivates optimizing the electrocatalyst price versus performance ratio using various approaches, with alloying of Pt with a less-expensive 3d transition metal (Pt-M; M = Cu, Ni, Fe, Co, etc.) being the closest to the production phase (Banham and Ye, 2017; Escudero-Escribano et al., 2018).
Not only do Pt-M alloys enable better utilization of Pt by diluting the core of the particles with a more abundant alloying metal (Stamenkovic et al., 2007a) but they also substantially enhance the ORR intrinsic activity by influencing the Pt surface structure-electronic properties through the welldocumented ligand and/or strain effect (Bu et al., 2016; Mezzavilla et al., 2016; Oezaslan et al., 2012a; Strasser et al., 2010).
We note that ensemble effects are also at play (Greeley et al., 2002), however not as widely discussed in the Pt-alloy ORR community.
These performance improvements of Pt alloying, however, come with an inherent price, namely the fact that the alloying transition metals (M) are thermodynamically unstable (Pourbaix, 1974) and readily dissolve from the electrode under acidic conditions inside the PEMFCs.
Pt-Mは熱力学的に不安定であるため、酸性雰囲気ではMが優先的に電極から溶出するという問題がある。
The dissolution of M can be slowed (but not completely stopped) by depleting the surface and near-surface region of Pt-M nanoparticles of M and thus forming a Pt-rich overlayer (Gatalo et al., 2019a).
Mの溶出を抑制するためには、表面表面や表面近傍の元素Mの量を減らすという方法がある。
This is referred to as the activation process, where M is dissolved either chemically by acid washing (Gatalo et al., 2019a) or electrochemically (Gatalo et al., 2019b) by for example in-situ electrochemical cycling.
元素Mを酸洗のような化学的もしくは電気化学サイクルによって溶出させる、いわゆる活性化と呼ばれるプロセスがある。
However, although this initial dissolution is highly desired, further dissolution of M can diminish its positive ligand and/or strain effect and result in a loss of ORR performance (Hodnik et al., 2014; Jovanovic et al., 2016; Papadias et al., 2018).
活性化処理によって元素Mの溶出は抑制されるが、元素Mによる配位効果やひずみ効果が抑制されることによってORR活性は低下する。
Furthermore, the dissolved M ions can also negatively affect the overall performance, especially in the membrane-electrode assembly (MEA)(Ahluwalia et al., 2018a; Braaten et al., 2019; Gasteiger et al., 2005; Mayrhofer et al., 2009).
溶出したMイオンは、特にMEAの性能に悪影響を及ぼす可能性がある。
One such example is the interaction between the dissolved M ions and Pt surface that blocks the active surface and thus affects its electrochemical performance.
溶出したMイオンがPt表面に近づき、活性表面における電気化学反応を妨害する。
In this sense, the most prominent example can be shown in the case of Pt-Cu alloy where Cu ions strongly interact, namely adsorb and reduce, on the Pt surface via well-known under-potential deposition (UPD); Cu can be found on both the cathode and the anode (Gatalo et al., 2019a, 2019b; Jia et al., 2013; Yu et al., 2012; Zhu et al., 2020).
Pt-Cuにおいて顕著:溶出したCuイオンがUPDとして知られている作用によってPt表面に吸着することにより、Pt表面における触媒反応を阻害する。
The second example is the replacement of protons in the ionomer with M cations, which results in higher O2 resistance as well as changes the water-uptake that consequently lowers the proton conductivity of the ionomer (Braaten et al., 2017, 2019).
もう1つの例は、Mカチオンがアイオノマーのプロトンと置換することによってアイオノマーのプロトン伝導度を下げることである。
This causes ohmic losses in the cell as well as slows the ORR reaction rate, as fewer protons are available at the electrode (Kienitz et al., 2011; Okada et al., 2002).
電池の抵抗損失やORR反応速度の低下につながる。
Lastly, any M present in the proton-exchange membrane will likely result in Fenton reactions and thus, its degradation that at the end leads to cell failure (Singh et al., 2018; Strlic et al., 2003).
どのMイオンもFe(II)と同様のフェントン反応によるヒドロキシラジカルの生成によりアイオノマーや電解質膜を劣化させる。
Consequently, despite the improvements in Pt utilization, intrinsic ORR activity, and overall electrocatalyst costs, the grand challenge of Pt-M electrocatalysts seems to be the long-term durability (Han et al., 2015).
初期性能は良くても、耐久性に問題が生じることもあるので、総合評価が必要である。
Therefore, if we wish to use Pt-M-containing electrocatalysts in PEMFCs, it is of
paramount importance to understand, lower, and eventually eliminate leaching of M. In order to do this, it is crucial for the PEMFC community to first understand the related chemical and electrochemical phenomena on a fundamental level. Many extensive studies have already addressed the structure-property behavior of Pt-based electrocatalysts (Baldizzone et al., 2015a; Gatalo et al., 2018; Gong et al., 2020; Han et al., 2015; Li et al., 2014; Oezaslan et al., 2012b; Strasser and Ku¨ hl, 2016; Strasser et al., 2010; Yu et al., 2012; Zalitis et al., 2020). However, most of these studies are based on the oversimplified, perfectly shaped model systems, which are based on either model single-crystal measurements or a result of purely theoretical calculations, and thus, do not cover the necessary complexity of real nanoparticulate systems behind these relations.
膨大な研究成果が蓄積されているが、問題が過度に単純化されていて、現実のナノ粒子系における複雑な過程をカバーできていない。
Upon looking at the structure-property of ‘‘real’’’ electrocatalysts, the opportunity to improve the Pt-M electrocatalysts still lies in optimizing their structure so that the activity improvement caused by M is the highest while minimizing the adverse effects of the leached M ions and thus, possible issues related to durability.
However, this task is tremendously complex.
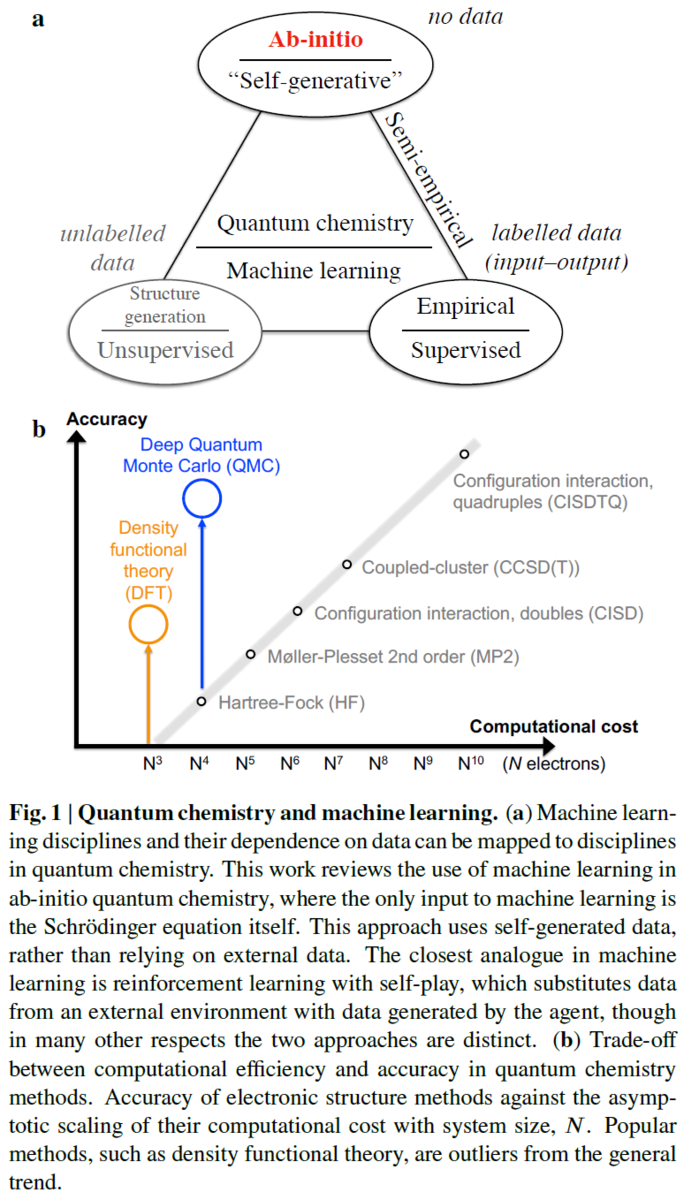
It is far from trivial to decide which structural feature to optimize in a given Pt-M system for the best overall performance, as real nanostructures are structurally far more diverse than the static model ones usually presented in the literature (for instance, pure Pt magic
number cuboctahedra in Figure 1 and Pt-alloy model core-shell nanoparticles in Scheme 1A) (Aarons et al., 2017).
Even for pure Pt surface, each atom has a certain coordination number (CN) that should affect its properties differently.
More specifically, the parameter of generalized coordination number (gCN), which
also takes the second sphere of neighbors into the account, was shown to directly correlate with ORR activity in the form of a volcano plot (Calle-Vallejo et al., 2015).
Interestingly, it was recognized that the concave kink sites with gCN of 8.1 are optimal and that the ORR activity of Pt nanoparticles with the size of approximately 2–4 nm can be enhanced by up to almost eight times merely by changing their shape (Figure 2) (Ru¨ ck et al., 2019).
On the other hand, Aarons et al., demonstrated that ‘‘real’’ nanoparticles (from
approximately 2 to 6 nm) contain a significantly different distribution of surface CNs with increased roughness (more defects) compared with the ideal cuboctahedral/truncated-octahedral nanoparticles (Figure 1).
This results in more sites with lower CN and thus higher O-binding ability that reduces the ORR activity.
In addition, they also observed that the amounts of active sites, with the optimal *OH-binding energy of 0.15–0.2 eV weaker than that of the Pt{111} surface (that is gCN is approximately 8.1 compared with 7.5 on Pt{111}), can vary for more than a factor of two at Pt particles with similar sizes.
When it comes to Pt nanoalloys, the degree of complexity that influences their activity and stability via the well-accepted structure-property relationships (Yang et al., 2017) becomes even higher.
Some examples of parameters increasing the complexity are chemical composition of the alloy (Gatalo et al., 2019c; Mezzavilla et al., 2016; Oezaslan et al., 2012a; Zalitis et al., 2020), degree of order in the crystal structure (intermetallic chemical order/disorder phases) (Xiong et al., 2019), the strain of surface Pt induced by, for instance, the thickness of Pt-shell, amount of retained less noble metal (Strasser et al., 2010) etc.
Many of them are interconnected and thus challenging to control.
Both extreme sensitivity and the complexity of Pt ORR electrocatalysts are directly addressed and elaborated in the recent viewpoint of Chattot et al. (Chattot et al., 2020).
In addition, besides the detailed atomic structure, which contains defects such as steps, adatoms, twinning, and dislocations, we must also consider the fact that the surface is not stagnant with the time under operation.
Therefore, the structure and thus property of each individual particle, which is far from the idealized models of spherical or cuboctahedral or truncated-octahedral core-shell particles (Scheme 1A), is also dynamically changing.
For this reason, dynamic alternations of nanostructures should be recognized as a structure-stability parameter, namely atomic movement and/or dissolution (Scheme 1B).
Looking at merely one TEM snapshot of nanoparticles atomic structure does not necessarily provide the only structure that induces certain function and also not a realistic idea of if and how specific atoms move or get dissolved (Scheme 1C).
On the other hand, when looking at the Pt-M electrocatalysts from the synthesis point of view, the most crucial step toward optimizing the structure and behavior of an electrocatalyst is to be able to tune its structural properties.
Indeed, colloidal chemistry recipes can produce nanoparticles with controlled shape by governing its growth via capping agents (Ahmadi et al., 1996; Neumann et al., 2017; Quinson et al., 2018; Speder et al., 2016; Xia et al., 2015).
However, besides issues related to the removal of this stabilizing molecules from the particles surface, it is a fact that it is practically impossible to synthesize a perfect model system that contains millions of ideal core-shell (spherical or cuboctahedra) nanoparticles with identical atomic structures, such as the one shown in Figure 1C and Scheme 1A.

A major challenge for Pt-M/C electrocatalysts is, besides shape, also optimizing the high-temperature thermal annealing treatments for each individual alloy to achieve the desired alloy phase (as defined in the phase diagrams).
In addition, each less-noble metal (as well as the Pt alloy itself) can also interact with the carbon support during annealing, as defined in the M-C phase diagrams, which further complicates the synthesis byforming thin, sometimes graphitized, carbon shells on the surface of the particles, depending on the nature of metal or alloy type (Baldizzone et al., 2014; Chong et al., 2018; Mezzavilla et al., 2016; Wang et al., 2019a; Xiao et al., 2019).
An additional step that is necessary for alloy-based electrocatalysts is activation, i.e. exposing the as-synthesized/thermally annealed material to appropriately corrosive chemical or electrochemical conditions (e.g. potential cycling activation; PCA (Gatalo et al., 2019b)) in order to remove M from the surface and near-surface region and thus reveal Pt active surface, namely Pt-shell.
The dealloying or leaching electrochemical phenomenon, of course, changes the structure of the nanoparticles depending on the initial shape, composition, crystal phase of the native alloy, and conditions (Pavlisic et al., 2016).
This process was shown to create some very active structures, referred to as stressed sites or surface distortion with the local disorder and straining (Chattot et al., 2018; Li et al., 2016).
At this point, one can ask an intriguing question whether the function of alloying Pt with unstable 3D metals is to create the superiorly active stressed sites with optimal gCNs (merely the atomic arrangement of Pt atoms in pure Pt nanostructures, as portrayed in Figures 1 and 2) or is it to alter Pt surface adsorption properties from underneath via strain and/or ligand effects (metal atoms below Pt surface in Pt-alloy nanostructures, as in Scheme 1A).
Overall, each Pt-M combination is uniquely defined by its properties and thus with its own set of constraints.
Therefore, one must understand their differences, which is again a very complex task.
These range from the synthesis (Gatalo et al., 2019c), alloys phase diagrams, metals mixing behavior (Dean et al., 2020), M-specific nanoalloys anisotropic inhomogeneity (Cui et al., 2013; Gan et al., 2014; Ruiz-Zepeda et al., 2019), standard redox potentials and/or Pourbaix diagrams of the less-noble metal, interaction of dissolved M ions from the electrolyte with Pt surfaces such as UPD (Gatalo et al., 2019b), pre-treatment (Baldizzone et al., 2015b; Gatalo et al., 2019a), general stability-dissolution-corrosion behavior (Gatalo et al., 2019b), and behavior in the PEMFC three-phase boundary environment (Ahluwalia et al., 2018a; Yu et al., 2012).
Lastly, there is currently no reliable database of as-synthesized shapes and their expected shape transformations (1) upon exposure to high temperatures of thermal annealing processes (Gan et al., 2016) and/or (2) upon activation (de-alloying) (Ruiz-Zepeda et al., 2019), much less a database that could take into the account such a wide range of
before-mentioned parameters.

As part of this work, we have tried to observe this structural complexity by investigating in total eight different commercially available Pt-based electrocatalysts, namely
(1) four Pt/C electrocatalysts (from Umicore, TKK, and JM) and
(2) four Pt-M/C electrocatalysts (M = Cu, Fe, Ni, Co; PK Catalyst; purchased at Fuel Cell Store).
In the first part, these electrocatalysts are initially systematically investigated using a rather conventional top-down approach with the purpose of exposing the differences of the seemingly comparable systems (Scheme 1).
Our structural investigation of these commercial electrocatalysts reveals a far more
complex picture, especially when compared with the ideal model systems.
The results suggest that it is practically impossible to control the synthesis of Pt-based nanoparticles in a way that would enable to fundamentally study and understand the structure-property behaviors as well as achieve major breakthroughs using a conventional top-down approach of studying such systems.
As a solution, we in the second part introduce a bottom-up approach by ‘‘playing the game’’ of ‘‘Spot the difference’’ (Scheme 2) (Hodnik and Cherevko, 2019) where an individual nanoparticle is studied at the atomic level before and after an electrochemical treatment.
By understanding the history of this specific nanoparticle and its changes at the atomic level, we gain indisputable evidence on some of the fundamental electrochemical phenomena and thus reliable structure-property relations.
This is, in our opinion, the only viable approach to study the structure-property relationships of Pt-based nanostructures.
In order to provide a proper comparison between both approaches (top-down and bottom-up), physical, chemical, and electrochemical properties were systematically characterized with a combination of classical characterization techniques.
These include X-ray diffraction (XRD), ex-situ transmission electron microscopy (TEM), thin-film rotating disc electrode (TF-RDE), as well as novel advanced characterization techniques such as highly sensitive online measurements of electrochemically dissolved metals (Pt and M; using electrochemical flow cell coupled to an inductively coupled plasma mass spectrometer; EFC-ICP-MS (Gatalo et al., 2016, 2019a, 2019b; Jovanovic et al., 2016, 2017a)) and detection of volatile compounds by direct coupling of an electrochemical cell to a mass spectrometer (EC-MS), which enables studying carbon corrosion via CO2 detection.
In addition, a newly developed modified floating electrode (MFE) methodology was used to enable identical location transmission electron microscopy (IL-TEM) with atomic resolution.
つづく