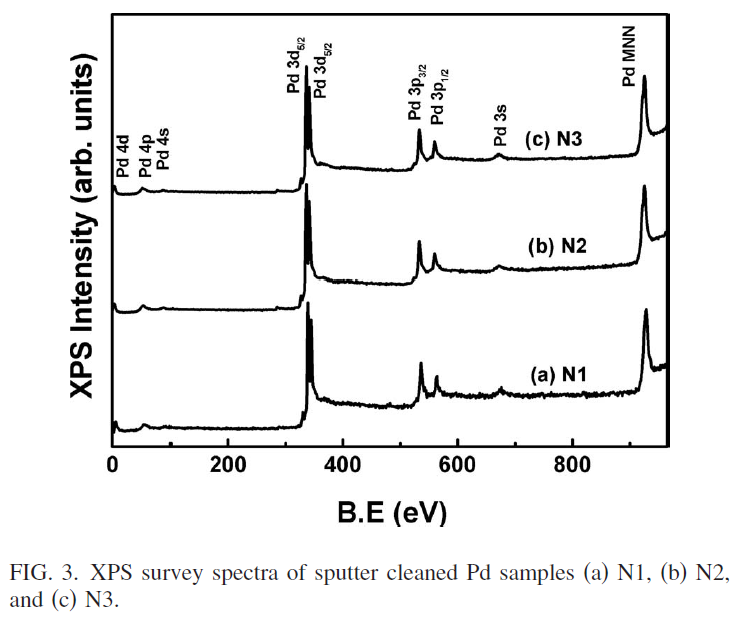
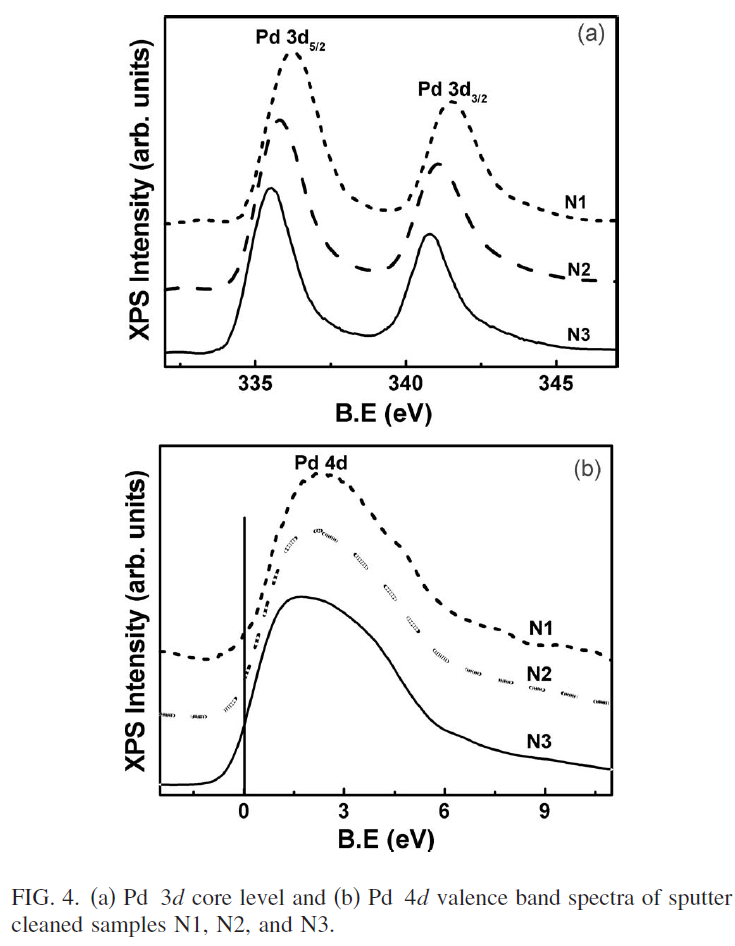
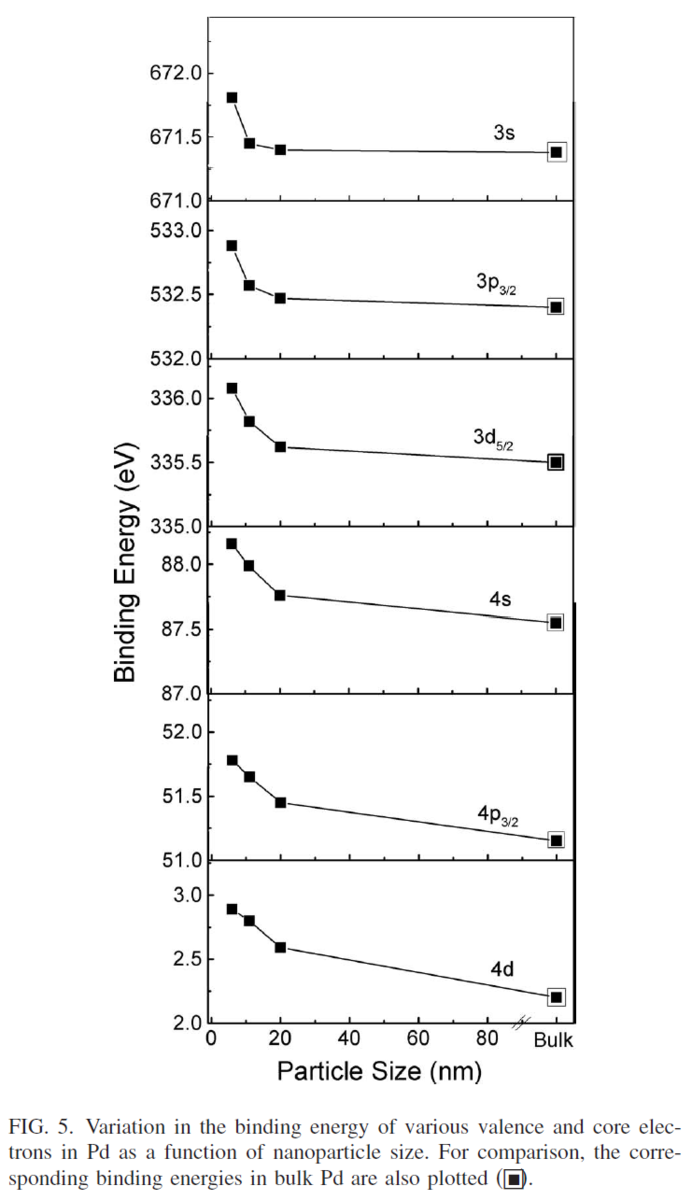
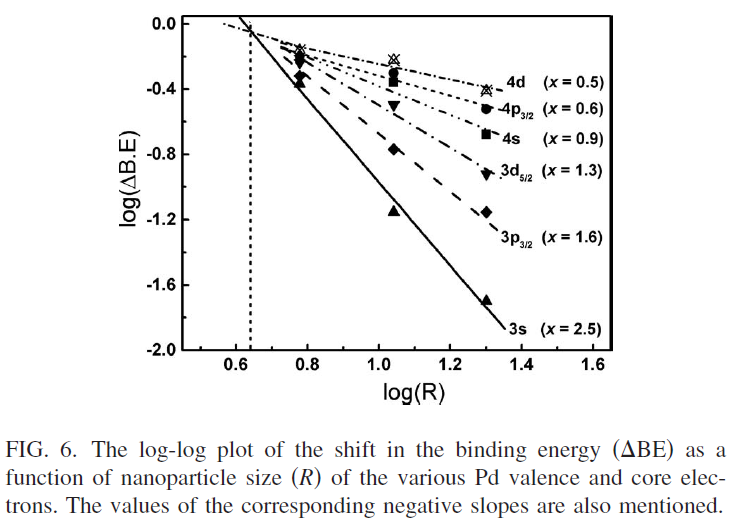
NMRが理解できないので、今日(2022年4月29日)からGW中に、理解に努めよう。
まずは、昨夜、NMRのテキストを2冊注文した。そのうちの1冊は日本分光学会から分光法シリーズとして出されている「NMR分光法」講談社である。
早速、いまだにぼんやりとしている縦緩和時間T1と横緩和時間T2の理解からはじめよう。まず、ムーアの本の約15行の説明を読み、次に、「NMR分光法」の2.4 核スピン緩和を読むことで、ようやく理解できた。(現象の物理的説明が詳しく書かれているので、じっくり読んでみる)
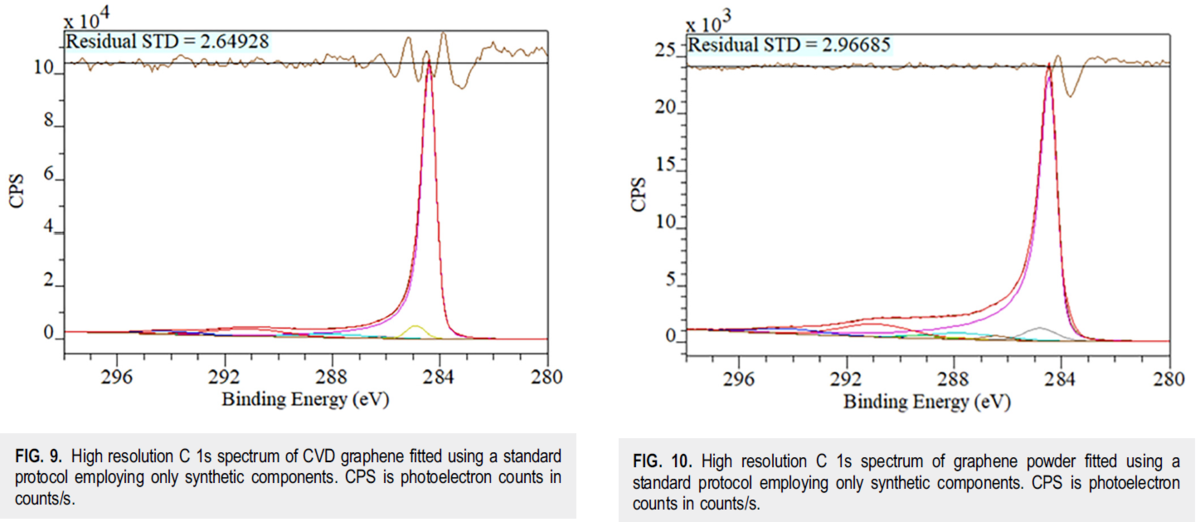
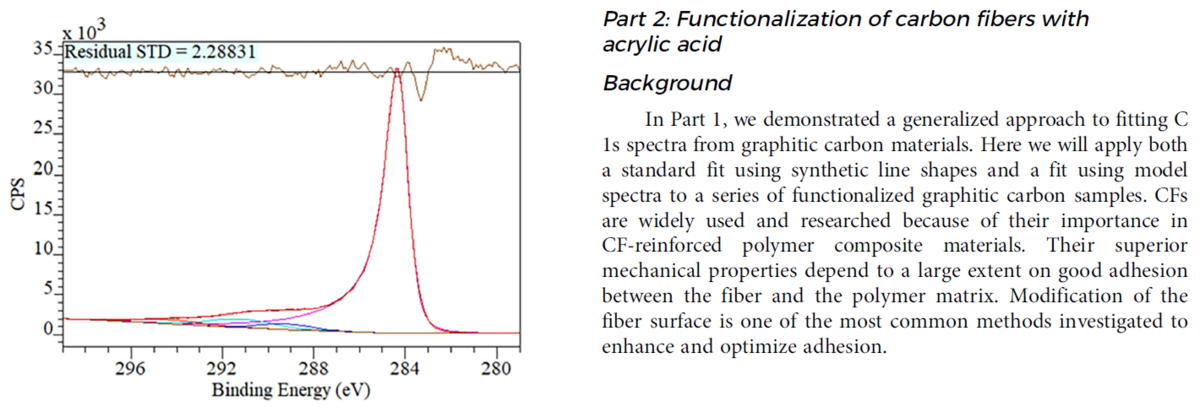
パルスNMRと高分解能NMRの計測データを解析できるようにしよう。
10月に勉強したのだが、やりなおし。
磁性とは何かをムーア先生に教えていただこう。
14章 分子構造と分子スペクトル
8. 磁気的性質
分子は永久磁気モーメントµと磁場によって誘起するモーメントαをもっている。
Bは磁気誘導、 H は磁場の強さ、 I は磁化の強さすなわち単位容積あたりの磁気モーメント
磁化率χは、I/Hで、負であれば反磁性、正であれば常磁性である。
9. 反磁性
磁場が加えられると、動いている電子の速度が変わり、Lentsの法則に従って、加えられた場と反対方向に働く磁場を生ずる。
原子中の電子の軌道が小さいため反磁性効果は小さいもので、グラム当たり、- 10^-6程度の大きさである。
10. 常磁性
常磁性を生ずる場合にはχは通常グラム当たり、10^-3 ~ 10^-4で、したがって小さな反磁性効果は問題にならない。
軌道角運動量の磁気モーメント
磁気回転比g
スピン角運動量
11. 磁性の型
12. 核の性質と分子構造
分子構造の実験的研究のまず目指すところは、分子内で原子核がどのような空間的配置をとるかということである。このような知見を得るには、構造内の結合間隔、結合角を知る必要がある。しかしこのような構造だけでは満足できるものではなく、電子が核の間でどのように分布するかを知る必要もある。電子の分布がわかると、結合の本性がはっきりし、最後には分子の化学反応性が説明できる。双極子モーメント、磁化率、スペクトル、X線ならびに電子線回折の測定をすれば電子構造についての知見が得られるが、これらの方法はすべて分子とある種の電磁場からなる外部検査器との相互作用に基づくものである。しかし大抵の場合、このような場は電子分布の微細な点まで明らかにするほど精密なものではない。
近年この分野において重要な進歩があった。その考えは核自体を検査器として用い、核をかこむ電子の分布を明らかにしようとするのである。核は無情な点電荷ではなくて、それが置かれた電気的な環境に敏感に反応する特性をもっているのである。核とその周囲との相互作用によって起こる効果を研究すると、分子内の電荷の分布の詳細がはっきりしてくることが多い。
核の重要な性質はその磁気モーメントと電気四重極モーメントである。核は固有の核スピンをもっており、したがって磁気モーメントµnをもつ小さな磁石として働く。核は固有の双極子モーメントはもたないが、四重極モーメントeQをもっている。核が四重極モーメントをもつとすれば、核の電荷分布は完全な球対称からずれていなくてはならない。このような核を普通回転楕円体で表すことができる。
13. 核の常磁性
陽子の磁気モーメント:2.79245核磁子
中性子の磁気モーメント:-1.9135核磁子:負に帯電した粒子のモーメントと同じような挙動をする。
14. 核磁気共鳴
系が低い状態に戻るのは、スペクトルを放出することによるだけではなく、緩和過程とよばれるいろいろな無放射機構によっても起こる。このような緩和過程が存在しないと、下の状態にいる方が上の状態にいるより少ないような熱平衡を維持する方法がないから、核磁気共鳴は実際に不可能になるだろう。
緩和機構には次の2種類のものがある。その1つは外部場の方向の核磁化がその平衡値に達しようとする緩和であって、縦の緩和(lomgitudinal relaxation)とよばれる。これは緩和速度が上の状態にある核の数の(平衡値からの)ずれの1乗に比例するから、1次反応式に従う。すなわち(8・11)式から次のようになる。
n - ne = (n - ne)0e-k1t = (n - ne)0e-t/T1
ここで速度定数k1の逆数は緩和時間T1、とよばれる。この過程はまたスピン-格子緩和ともよばれ、配向した核のまわりの物体中のいろいろな変動する局所場によるものである。多くの機構のうちの一例として、常磁性イオンが水に付着すると、イオンの不対電子の強い磁場のため陽子の緩和時間T1が非常に減少することが見出されている。図14・11には水中の三つのイオンについてこの効果が示されている。
第2番目の緩和過程は横の緩和(transverse relaxation)(T2) とよばれる。場の方向のまわりを歳差運動をしている核が互いに同一位相にあると、磁場の軸 Z に垂直な XY 面に磁気モーメントの正味の成分が残ることになる。したがってこの位相を破壊するような何らかの場があると、磁気モーメントの XY 成分の緩和を起すことになろう。このような過程の一つはスピンースピン緩和であって、スピンの高い状態の核がスピンを交換して隣の原子核へエネルギーを移すのである。
15. 化学シフトとスピンスピン分裂
核の周囲もまた核が感知できる程度の小さな場の効果をもつことができる。このことのために分子構造や化学結合の本性を研究する際 NMR が非常に有用になる。核のまわりの電子は外部場の作用をうけて誘起反磁性を生じ、部分的に核をしゃへいする。このしゃへい効果は外部場の約100万分の10にすぎないが、NMR 測定の精度は非常によくて、この効果は1%以内の精度で容易に測定される。この結果は化学シフト(chemical shift)とよばれる。図14・10では、CH3CH2OHの陽子共鳴がこの構造の三つの異なった種類のHでそれぞれいくらか違っている例を見た。化学シフトは外部場によって誘起される反磁性によるものであるから、その絶対値は外部場の強さによって決まる。
図14・10(a)に示した NMR スペクトルをさらに分解能のよい装置で調べると、CH2のピークは四つの線に、CH3のピークは3つの線に分裂する。高分解能で得られたスペクトルを図14・10(b)に示す。CH2やCH3のピークを分裂した効果は、核のまわりの電子が核をしゃへいしたため生ずる化学シフトではない。その理由は、観測された分裂が加えた場の強さによらないからである。この効果は一組の陽子の核スピン(磁気モーメント)と他の組のものとの相互作用によって生ずるものである。したがってスピンースピン分裂(spin-spin splitting)とよばれる。
以下に簡潔な説明がなされているが、図示できないので省略する。
昨日購入した「NMR分光法」においては、スピンースピン結合として、12ページ以上にわたって詳細に説明されているが、難しく書かれていて、よく理解できなかった。
**********2022年4月29日再開