白金と酸素
白金の酸化、酸化白金の還元、酸素の吸着、酸素の脱着、燃料電池のアノード反応とカソード反応、燃料電池のカソードにおける酸素還元反応(ORR)などをテーマとする。
[50]:Origin of the Overpotential for Oxygen Reduction at a Fuel-Cell Cathode
J. K. Nørskov et al., J. Phys. Chem. B, 108, (2004) 17886

2022/10/15:この図Figure 4の説明文を見てみよう。
The activities constructed from Table 2 are plotted in Figure 4 as a function of the O binding energy and in Figure 5 as a function of both the O and the OH binding energies. A nice volcano appears. In good agreement with experiment, it shows that Pt and Pd are the best catalysts for oxygen reduction.

Figure 5では、OだけでなくOHの結合エネルギーと活性との相関性も高いことが示されている。
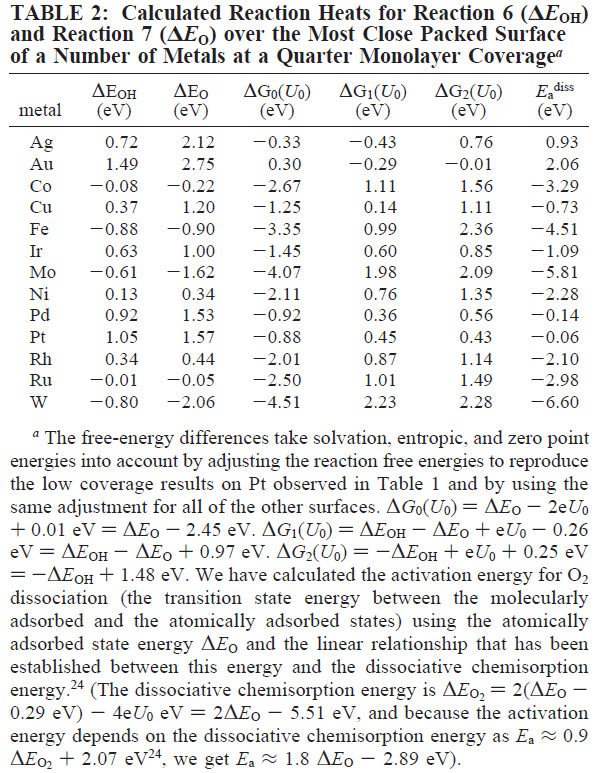
結合エネルギーΔE0は、Auのように酸化されにくい元素は正で大きな値、Feのように酸化されやすい元素は負で大きな値を示している。
この結合エネルギーΔE0は、金属表面における次の反応式から得られる*とO、*とOHの結合エネルギーということのようである。*は、金属表面の吸着サイトのことである。
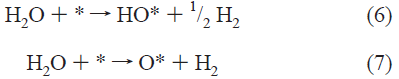
吸着サイトの原子配列や電子構造によって結合エネルギーΔE0が変化し、活性も異なる。Fig. 4やFig. 5から、Ptよりもプラス方向にΔE0をシフトさせることができれば活性の向上が期待されるということで、このプロットは触媒粒子の設計の指針になるということで形を変えて今でも使われている(ようである)。
この方向で極めようとすると、表面原子配列や合金化、規則化/不規則化などに首をつっこむことになるが、それについては、計算機実験を繰り返しているグループがある(らしい)。
ここでは、論文から離れて、標準電極電位の観点から、Fig. 4, 5にプロットされている元素を追跡してみよう。
Fig. 4では、マイナス側から、W, Mo, Fe, Co, Ru, Ni, Rh, Cu, Ir, Pd, Pt, Ag, Auの順である。これに対して、ウイキペディアで調べた標準電極電位の結果を以下に示す。
| Fe | Fe2+ + 2 e− |
⇌ | Fe(s) | -0.44 |
| Co | Co2+ + 2 e− |
⇌ | Co(s) |
-0.28 |
| Ni | Ni2+ + 2 e− |
⇌ | Ni(s) | -0.25 |
| Cu | Cu2+ + 2 e− |
⇌ | Cu(s) | 0.337 |
| Ru | Ru2+ (aq) + 2 e− |
⇌ | Ru | 0.455 |
| Ag | Ag+ + e− |
⇌ | Ag(s) | 0.7996 |
| Pd | Pd2+ + 2 e− |
⇌ | Pd(s) | 0.915 |
| Pt | Pt2+ + 2 e− |
⇌ | Pt(s) | 1.188 |
| Au | Au3+ + 3 e− |
⇌ | Au(s) | 1.52 |
| Au | Au+ + e− |
⇌ | Au(s) | 1.83 |
Introduction
Low-temperature fuel cells are attracting considerable interest as a means of producing electricity by direct electrochemical conversion of hydrogen and oxygen into water.
There are, however, severe shortcomings of the present technology, which need to be overcome to make low-temperature fuel cells more economically attractive.
One of the most important problems is related to the low rate of the cathode reaction where oxygen is reduced
![]()
Pt is the commonly used electrode material, but there is a considerable overpotential associated with this reaction over Pt. For some reason, the kinetics of the cathode reaction make it much slower than the anode reaction,
![]()
and there is presently no consensus why this is so.
In the following, we use density functional theory (DFT) calculations to gain some insight into the cathode reactions.
DFT calculations can provide information about the stability of surface intermediates in the reactions, which cannot be easily obtained by other means.
We start by considering the simplest possible reaction mechanism over a Pt(111) surface.
We introduce a method for calculating the free energy of all intermediates as a function of the electrode potential directly from density functional theory calculations of adsorption energies for the surface intermediates.
On this basis, we establish an overview of the thermodynamics of the cathode reaction as a function of voltage, and we show that the overpotential of the reaction can be linked directly to the proton and electron transfer to adsorbed oxygen or hydroxide being strongly bonded to the surface at the electrode potential where the overall cathode reaction is at equilibrium.
We introduce a database of density functional theory calculations of energies of the surface intermediates for a number of metals and show that, on this basis, we can establish trends in the thermodynamic limitations for all the metals in question.
The model predicts a volcano-shaped relationship between the rate of the cathode reaction and the oxygen adsorption energy.
The model explains why Pt is the best elemental cathode material and why alloying can be used to improve its performance.
The Simplest Model
To introduce the basic concepts, we first study the simple dissociative mechanism Oxygen Reduction at a Fuel-Cell Cathode
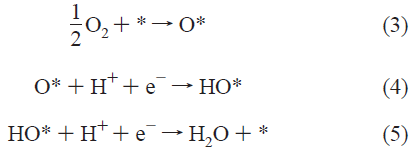
Here, * denotes a site on the surface.
式(3)は、酸素が触媒表面に化学吸着する反応
Later, we will also discuss in detail the associative mechanism where O2 does not dissociate before it is hydrogenated, and we will show that although this changes several important details of the reaction kinetics, it does not affect the main conclusions, in particular, regarding the overall variations in the reaction rate from one metal to the next.
The stability of the intermediates O* and HO* can be calculated on a Pt(111) surface.
In Table 1, we show the calculated binding energies defined as the reaction energies of the reactions
where H2O and H2 are in the gas phase.
The stability of both adsorbed OH and adsorbed O depends strongly on the oxygen
coverage; therefore in Table 1, we include results for two different oxygen coverages to illustrate this effect.
We now introduce our procedure for calculating the free energy of the intermediates of the electrochemical reactions (eqs 3-5). It goes in six steps:
1. By setting the reference potential to be that of the standard hydrogen electrode, we can relate the chemical potential (the free energy per H) for the reaction (H+ + e-) to that of 1/2H2 (eq 2 is in equilibrium).
![]()
This means that, at pH = 0 in the electrolyte and 1 bar of H2 in the gas phase at 298 K, the reaction free energies of eqs 6 and 7 are equal to those of the reverse reactions eq 5 and eq 4 + 5 at an electrode potential of U = 0 relative to the standard hydrogen electrode.
2. To model the water environment of the electrochemical cell, we include the effect of a monolayer of water on the stability of adsorbed O and OH in the calculation.
For the low coverage results, we have simply added water to fill the surface, and we have added bilayer of water on top of the adsorbed O and OH for the high coverage results as proposed by Ogasawara et al.
The interaction with water stabilizes OH groups on the surface relative to adsorbed oxygen due to hydrogen bonding.
表1のOHの結合エネルギーにおいて、ΔEよりΔEw,waterが小さくなっている。
The effect of the water layer on adsorped oxygen is negligible.
表1の酸素の結合エネルギーにおいて、ΔEとΔEw,waterが等しい。
3. We include the effect of a bias on all states involving an electron in the electrode, by shifting the energy of this state by -eU, where U is the electrode potential.
4. The adsorbed states also interact with the field set up outside the surface by the electrochemical double layer.
The most rigorous treatment would involve a detailed model of the water and two electrodes and the electrolytes with a bias.
This would entail a calculation for a nonequilibrium system with two Fermi levels, which is not currently possible.
A simple estimate of the field effect can be obtained by calculating the coupling
between the dipole moment of the adsorbed state and the average field just outside the surface.
For O* and OH*, this gives a small effect because the dipole moments are small, 0.035 and 0.05 eÅ, respectively, on Pt(111).
At a bias of 1 V relative to the point of zero charge, the typical average field is ~0.3 V/Å, assuming the width of the double layer to be ~3 Å.
The effect of the electrical field on the adsorption energy is thus approximately 0.05 eÅ x 0.3 V/Å = 0.015 eV.
We neglect this in the following.
5. At a pH different from 0, we can correct the free energy of H+ ions by the concentration dependence of the entropy: G(pH) = -kT ln[H+]= kT ln 10 pH.
6. We calculate free energies of the intermediates at zero potential and pH = 0 as ΔG = ΔEw,water + ΔZPE - TΔS, where ΔE is the reaction energy of eq 6 or 7, ΔZPE is the difference in zero point energies due to the reaction, and ΔS is the change in entropy.
All of the parameters have been taken from DFT calculations or standard tables for gas-phase molecules and are shown in Appendix 1.
The electronic structure problem has been solved using density functional theory in a plane wave pseudopotential implementation, employing the ultra-soft pseudopotentials of Vanderbilt to represent the ionic cores.
All calculations were performed with the RPBE exchange-correlation functional on periodically repeated metal slabs. RPBEは交換相関汎関数の1つである。
The Pt calculations were done on a (3 x 2) three-layer fcc(111) slab at the RPBE lattice constant of Pt (4.02 Å) separated by at least five equivalent layers of vacuum.
The bottom two layers were fixed, and the top layer was allowed to relax.
A 3 x 4 x 1 Monkhorst-Pack k-point sampling was used.
The plane wave cutoff was 340 eV, and the density was treated on a grid corresponding to a plane wave cutoff at 500 eV.
For the results presented in Table 2, the OH adsorption energies were calculated on (2 x 2) four-layer slabs with the top two layers relaxed. A 4 x 4 x 1 Monkhorst- Pack k-point sampling was used, with maximum symmetry applied to reduce the number of k points in the calculations.
The dipole correction was used in all cases.
The plane wave cutoff was 340 eV for OH, 350 eV for H, and 450 eV for the O adsorption calculations.

表1を理解できたかどうか確認してみよう。6つのステップに分けて説明されているようだが、ぼんやりしたままである。
次は表2をみてみよう。
Observing the oxidation of platinum
Matthijs A. van Spronsen, Joost W.M. Frenken & Irene M.N. Groot Nature Communications volume 8, Article number: 429 (2017)
PEFCのカソードにおけるORR反応を理解するには、白金の酸化について理解を深めることが重要ではないかと思い、platinum oxidationで検索した結果、この論文に行き当たった。
J. K. Nørskovらのvolcano plotの横軸は酸素結合エネルギーである。すなわち酸素が触媒表面に化学吸着する反応のエネルギーである。すなわち金属表面における金属と酸素の結合エネルギーである。白金の場合は、白金と酸素の結合エネルギーである。漠然と、白金は酸化されないと思っていて、白金酸化物は存在しないだろうと思っていた。常温の大気中では目に見えるような酸化物は生じていないようである。
ウィキペディアで白金を調べると、白金は酸化されにくいとしか書かれておらず、酸化白金やPtO2の記述はない。
英語バージョンでは、酸化白金について説明されている。英語バージョンの一部を和訳すると、プラチナは耐腐食性に優れています。バルク プラチナは空気中でどの温度でも酸化しませんが、約 400 °C に加熱することで簡単に除去できるPtO2の薄い表面フィルムを形成します。酸化白金(IV)、PtO2は、「アダムス触媒」としても知られ、水酸化カリウム(KOH) 溶液および濃酸に溶解する黒色の粉末です。PtO2とあまり一般的でないPtOはどちらも加熱すると分解する。
For the Pt-based automotive catalyst, the most essential model is the Pt(111) single-crystal surface. This surface has the lowest surface energy and is expected to form the
largest facets in a real catalyst. Pt(111)は表面エネルギーが最小であるから最大のファセット面を持つことが期待される。
The interaction of O2 with Pt(111) has been extensively studied under traditional surface science conditions, i.e., ultra-high vacuum (UHV). It was found that O2 binds molecularly
below 160 K, above which it dissociates readily and forms a p(2 × 2)-O chemisorption overlayer with a saturation coverage of 0.25 ML. 超高真空中で160 K以下では、分子吸着し、それ以上の温度では p(2 × 2)-O 原子吸着し、飽和被覆率は0.25である。
High-temperature exposure or exposure to stronger oxidants, such as NO2, O3, and atomic oxygen, was needed to create higher O coverages.
高温でより強い酸化剤に晒すと、これ以上の被覆率となる。
This included a surface oxide consisting of one-dimensional (1D) oxidic rows, which were
forming honeycomb-like superstructures. Using these harsh conditions, even PtO2 could be created. 厳しい酸化条件にすればPtO2でさえも生じる可能性がある。
There is no guarantee that the structure of a catalyst observed in UHV is the same as the structure present under reaction conditions. UHVではなく現実の反応(動作)条件での構造を調べる必要がある。
This structure can only be elucidated when it is probed in situ, i.e., under high-pressure and elevated-temperature conditions. この構造は高温高圧でその場観察しないと明らかにはならない。
Two independent in situ surface X-ray diffraction (SXRD) studies showed the formation of bulk-like α-PtO2. These observations were contradicted by a near-ambient-pressure
(NAP) X-ray photoelectron spectroscopy (XPS) study, which showed the formation of a surface oxide at similar temperatures as in the SXRD experiments, but at lower pressures. This surface oxide was found to be an intermediate in the bulk oxidation of Pt, which only started at much higher temperatures. In a recent NAP XPS study, it was found that prolonged exposure to oxidizing conditions was needed to form Pt oxide.
SXRDとNAP XPSとではPtO2が観察される条件は若干異なるようである。先に進もう。
The most important questions remain unanswered. What is the structure formed under catalytically relevant conditions? If it is an oxide, is this a surface or bulk oxide?
PtO2が形成されたときどのような構造になるのか。
In this work, the oxidation of Pt(111) is probed with O2 pressures of 1–5 bar and at 300–538 K using in situ scanning tunneling microscopy (STM). Interestingly, the formation of
α-PtO2 is not observed, instead two stable surface oxides form. The first has a structure in which equilateral triangles are arranged into spoked wheels. The lattice constant within the spokes is close to that of α-PtO2. The second structure consist of a pattern of rows which are lifted from the surface and consisted of nearly half the amount of Pt atoms in the top layer. These surface oxide are not stable without the high O2 pressure indicating
that the O atoms in these structures are very reactive, making them relevant for catalysis.
PEFCの動作条件よりかなり厳しい条件でもα-PtO2は生じないということで先に進む。
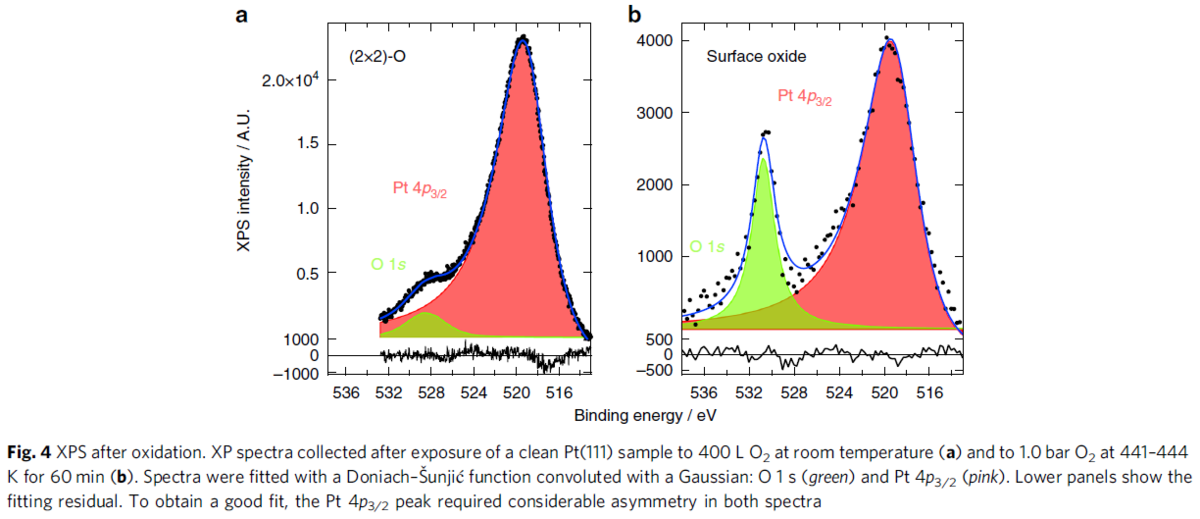
この論文は、排ガス用の酸化触媒なので、高温条件でのPtの酸化について調べているが、PEFCは前提条件が異なるのでここまでとする。常温、UHV、1 barでは、酸素ガスはPt(111)表面で原子(解離)吸着し、被覆率が0.25で飽和するというのは参考情報だとしても興味深いし、上記のXPSスペクトルも参考になる。
