基礎からの燃料電池触媒:2021年10月3日~
山梨大学の燃料電池ナノ材料研究センターのパンフレットによれば、1978年4月に工学部付属燃料電池実験施設を設置しており、1989年4月には学内特別施設電気化学エネルギー変換研究室を設置、2001年4月にはクリーンエネルギー研究センターを設置、そして、2008年4月に当該センターが設置されている。ということで、目に見える形で燃料電池にフォーカスしてからでもすでに40年以上の歴史を有している。
当該センターとNEDOとの関係:「本学では、2008年度から2014年度にNEDOのHi-PerFCプロジェクト(※1)を受託し、2015年度から2019年度にNEDOのSPer-FCプロジェクト(※2)を受託し、燃料電池の高出力化、高耐久化、高効率化に資する触媒や電解質材料およびそれらの性能を極限まで発揮させる触媒層の研究に取り組み、世界でも注目される多くの成果を挙げてまいりました。」
2020年度からは、「この度、新たにNEDOからECCEED’30-FCプロジェクト(※3) ECCEED’40-FCプロジェクト(※4)を受託しました。2020年度からこれまでの成果を活かしながら新たな発想を取り入れることにより、NEDO技術マップ等で定められるシナリオに基づき、高効率、高耐久、低コストの燃料電池システムを実現するための技術を開発します。」
ということで、燃料電池の研究開発の現状と展望を知るためには、当該センターに出かけて行ってお話を聞くのが良さそうである。できるだけ早い機会に訪問したいと思う。
今日のテーマは過電圧:
燃料電池のカソードにおける酸素還元反応に対する過電圧の起源に関する論文を読んでみよう。2004年の有名な論文のようだ。
J. K. Nørskov et al., Origin of the Overpotential for Oxygen Reduction at a Fuel-Cell Cathode, J. Phys. Chem. B, 108, 17886 (2004)
水素と酸素から水を生じる電気化学変換反応を利用する低温燃料電池において、酸素を還元するカソード反応が遅いということは、大きな問題点の1つである。それは、水素を酸化するアノード反応よりも遅いのだが、なぜそうなのかについてはコンセンサスが得られていない。
In the following, we use density functional theory (DFT) calculations to gain some insight into the cathode reactions.
DFT calculations can provide information about the stability of surface intermediates in the reactions, which cannot be easily obtained by other means.
We start by considering the simplest possible reaction mechanism over a Pt(111) surface.
We introduce a method for calculating the free energy of all intermediates as a function of the electrode potential directly from density functional theory calculations of adsorption energies for the surface intermediates.
On this basis, we establish an overview of the thermodynamics of the cathode reaction as a function of voltage, and we show that the overpotential of the reaction can be linked directly to the proton and electron transfer to adsorbed oxygen or hydroxide being strongly bonded to the surface at the electrode potential where the overall cathode reaction is at equilibrium.
We introduce a database of density functional theory calculations of energies of the surface intermediates for a number of metals and show that, on this basis, we can establish trends in the thermodynamic limitations for all the metals in question.
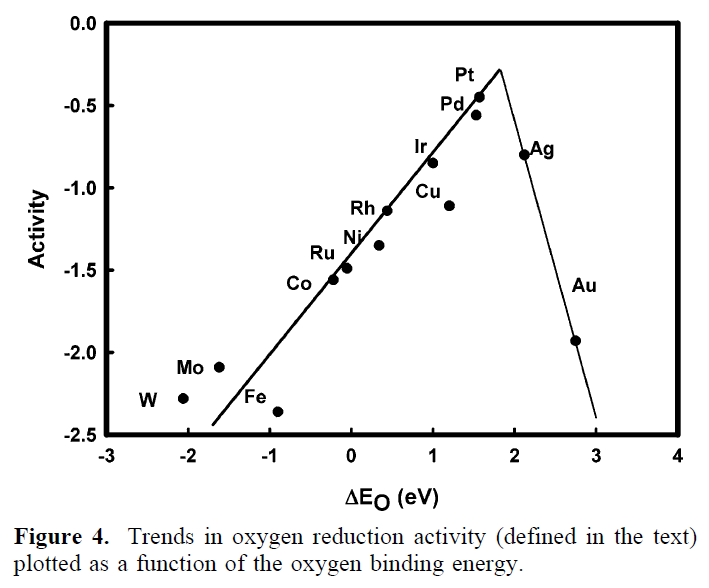
The model predicts a volcano-shaped relationship between the rate of the cathode reaction and the oxygen adsorption energy.
The model explains why Pt is the best elemental cathode material and why alloying can be used to improve its performance.
***残念だが、ブログを中断する***
