燃料電池と機械学習(Ⅱ)(fuel cell and machine learning part 2):2021年9月
"deep learning fuel cell"で検索した文献をランダムに読んでみる。⇒ 白金電極上の酸素の還元反応のメカニズムを原子レベルで解明するために必要な、反応中間体の吸着エネルギーや自由エネルギーの計算精度を上げるために、吸着物化学環境ベースグラフコンボリューションニューラルネットワークが考え出され、その計算結果を基に反応機構を解明しようとしている論文を読む。
9月1日(水)
Physics-constrained deep learning for data assimilation of subsurface transport
Haiyi Wu and Rui Qiao, Energy and AI 3 (2021) 100044
a b s t r a c t Data assimilation of subsurface transport is important in many energy and environmental applications, but its solution is typically challenging. In this work, we build physics-constrained deep learning models to predict the full-scale hydraulic conductivity, hydraulic head, and concentration fields in porous media from sparse measure- ment of these observables. The model is developed based on convolutional neural networks with the encoding- decoding process. The model is trained by minimizing a loss function that incorporates residuals of governing equations of subsurface transport instead of using labeled data. Once trained, the model predicts the unknown conductivity, hydraulic head, and concentration fields with an average relative error < 10% when the data of these observables is available at 12.2% of the grid points in the porous media. The model has a robust predictive performance for porous media with different conductivities and transport under different Péclet number (0.5 < Pe < 500). We also quantify the predictive uncertainty of the model and evaluate the reliability of its prediction by incorporating a variational parameter into the model.
データ同化(Data assimilation)は、理論(通常は数値モデルの形式)と観測値を最適に組み合わせようとする数学的分野です。たとえば、システムの最適な状態推定を決定する、数値予測モデルの初期条件を決定する、観測されているシステムの(物理的な)知識を使用してまばらな観測データを補間するなど、さまざまな目標が求められる場合があります。観測データに基づいて数値モデルパラメータをトレーニングします。目標に応じて、さまざまな解決方法を使用できます。データ同化は、分析対象のシステムの動的モデルを利用するという点で、他の形式の機械学習、画像分析、統計的手法とは異なります。by ウイキペディア
ペクレ数(ペクレすう、英: Péclet number、Pe)は、連続体の輸送現象に関する無次元数。この名はフランスの物理学者Jean Claude Eugène Pécletにちなむ。流れによる物理量の移流速度の、適切な勾配により駆動される同じ量の拡散速度に対する比率と定義される。物質移動の文脈では、ペクレ数はレイノルズ数とシュミット数の積である。熱流体の文脈では、熱ペクレ数はレイノルズ数とプラントル数の積に相当する。by ウイキペディア
1. Introduction
Heterogeneous porous media are ubiquitous in natural and engineering systems. Determining their transport properties and the transport of fluids and solutes in them are important in many energy applications. For example, in PEM fuel cells, the flow in the gas diffusion layers and mass transfer in the proton-conducting membrane play a key role in controlling their performance and thus must be predicted accurately in cell design [ 1 , 2 ]. In oil recovery, the distribution of permeability in highly heterogeneous oil reservoirs governs oil recovery and predicting oil transport in them is essential for designing oil recovery strategies [ 3 , 4 ]. This is especially true when CO 2 injection is used to enhance oil recovery [ 4 , 5 ]. Classical methods for solving transport in porous media require full knowledge of transport properties of porous media (e.g., hydraulic conductivity) as well as the initial and boundary conditions [6] . It is, however, challenging to obtain highly resolved transport properties of porous media, especially in the presence of high spatial heterogeneity [ 7 , 8 ]. Without such highly resolved data, predicting the transport in porous media is challenging.
Enhanced oil recovery (abbreviated EOR), also called tertiary recovery, is the extraction of crude oil from an oil field that cannot be extracted otherwise. EOR can extract 30% to 60% or more of a reservoir's oil,[1] compared to 20% to 40% using primary and secondary recovery.[2][3] According to the US Department of Energy, carbon dioxide and water are injected along with one of three EOR techniques: thermal injection, gas injection, and chemical injection.[1] More advanced, speculative EOR techniques are sometimes called quaternary recovery.
ナノレベルの空孔中の物質輸送に対して、油田における二酸化炭素注入による油の回収というマクロレベルにおける物質輸送と対比させているとことが面白い。
Data assimilation can be an effective method for predicting full-scale data (e.g., transport properties of porous media and transport behavior in them) from sparse measurements.
Data assimilation is a process that seeks to combine physical theory and observed data to estimate the state of a system or to interpolate sparse observation data using physical theories.
Data assimilation has been used to reconstruct the observed history of atmosphere data [9] and to resolve difficulties of parameter estimation and system identification in hydrologic modeling [10].
However, traditional data assimilation methods for solving the transport in porous media can be computationally expensive because of the high heterogeneity in many porous media and the highly nonlinear equations governing the transport behavior.
ここでDeep Learningの登場!
Deep learning-based methods can potentially tackle the above challenges. They have shown promise in solving forward and inverse transport problems in complex systems [11-15]. For instance, deep convolutional encoder-decoder networks have been used to predict the distribution of thermal conductivity in composites using sparse temperature measurements [15]. Surrogate models based on physics-constrained deep learning has been used for uncertainty quantification of flow in stochastic media [ 16 , 17 ]. Recently, physics-informed neural networks (PINNs) were developed to solve partial differential equations with sparse measurement data as input [ 18 , 19 ]. PINNs-derived models have been used for data assimilation in subsurface transport and the accuracy of these models working with different input measurements has been carefully studied [20] . These pioneering studies point to exciting opportunities of using deep learning in data assimilation.
In this work, we build physics-constrained deep learning models to solve a data assimilation problem in porous media. Specifically, we focus on subsurface fluid and solute transport in the presence of heterogeneity in hydraulic conductivity. Deep learning models are developed to predict full-scale hydraulic conductivity, hydraulic head, and solute concentration from sparse measurements of these observables. While we focus on data assimilation of subsurface transport in the presence of heterogeneity in hydraulic conductivity, which is similar to the subject in Ref. [20], the machine learning models we used are very different. The DNN model in Ref. [20] is mainly based on physics-informed neural networks (PINNs), which were developed to solve partial differential equations with sparse measurement data as input [ 18 , 19 ]. It is useful to note that PINNs-based models are built with several fully connected neural layers that involve a large set of learning parameters, and some models do not yet provide information on the uncertainty and reliability of their predictions. In this work, instead of using fully connected neural layers, we adopt convolutional neural networks, which often result in a smaller number of learning parameters easier for training than the fully connected neural networks. We also explore the possibility of gauging the uncertainty and reliability of the model prediction by introducing a variational parameter into the deep learning model. The developed models are trained using sparse measurement data by minimizing the residuals of governing transport equations and the loss due to mismatch between predicted and measured data at measurement points. The performance of the models is investigated under different conductivity fields, nature of solute transport, and the noise level of input measurement.
2. Problem definition
これをフォローするのは、まだ、難しい。
Without losing generality, we consider the subsurface transport in a two-dimensional (2D) square-shaped porous domain Ω∈[ 0 , 1 ] ×[ 0 , 1 ] at steady state. Fluid flow is described by the Darcy model:
ダルシーの法則は、多孔質媒体を通る流体の流れを表す方程式です。この法則は、地球科学の一分野である水文地質学の基礎を形成する、砂床を通る水の流れに関する実験[1]の結果に基づいて、ヘンリー・ダルシーによって策定されました。byウイキペディア
3. Physics-constrained deep learning model
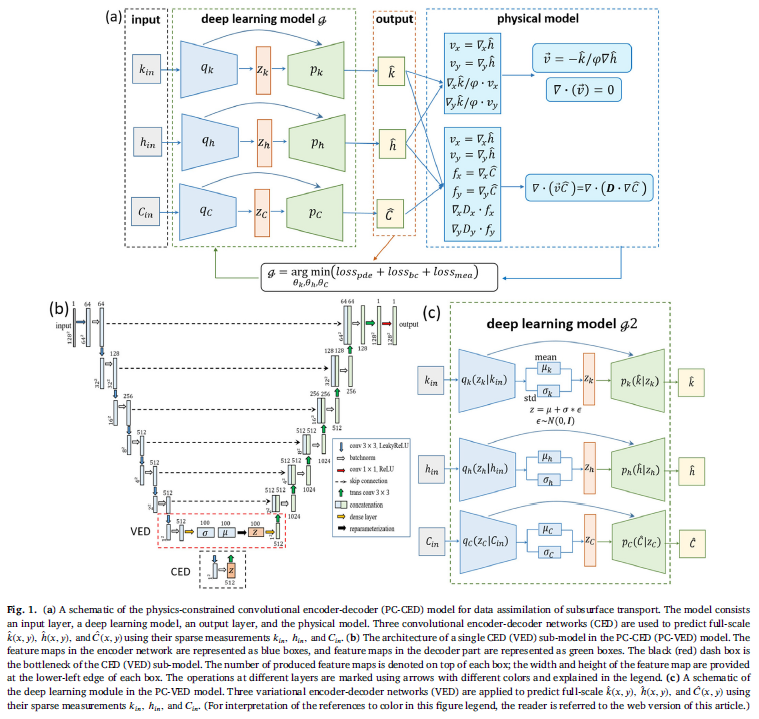
We use deterministic and probabilistic deep learning models to solve the data assimilation problem defined above. All the reference data in this work are numerical data. The deterministic model is based on physics-constrained convolutional encoder-decoder networks (PC-CED). There are three main parts in a PC-CED model: an encoder network, a latent space, and a decoder network. The encoder network takes the sparse measurement data ℎ 𝑖𝑛 , 𝑘 𝑖𝑛 , 𝐶 𝑖𝑛 as input and is trained to compress and extract important features and correlations from the input data. The extracted features have a much lower dimension than the input features and are stored in the latent space. The decoder network then projects the low-dimensional features in the latent space to high-dimensional space to predict the full-scale data k ( x,y ) , h ( x,y ) , C ( x,y ).
9月2日(木)
Fundamentals, materials, and machine learning of polymer electrolyte membrane cell technology、の表12に掲載されている機械学習関連のツールや種々のデータベースを紹介しているウェブサイトについて調べてみる。
Table 12 : Publicly accessible professional machine-learning tools for chemistry and material, and structure and property databases for molecules and solids. The table is developed following format of that in Ref.[224] by adding additional information.
Machine learning tools for chemistry and material:Amp, ANI, COMBO, DeepChem, GAP, MatMiner, NOMAD, PROPhet, TensorMol,
Computed structure and property databases:AFLOWLIB, Computational Materials Repository, GDB, Harvard Clean Energy Project, NOMAD, Open Quantum Materials Database, NREL Materials Database, TEDesignLab, ZINC
Experimental structure and property databases:ChEMBL, ChemSpider, Citrination, Crystallography Open Database, CSD, ICSD, MatNavi, MatWeb, NIST Chemistry WebBook, NIST Materials Data Repository, PubChem
ANI:Works only under Ubuntu variants of Linux with a NVIDIA GPUと書かれていて、Ubuntuを使うことが前提となっている。Windowsでは動かないので、現時点では、少しハードルが高い。
REQUIREMENTS: Python 3.6 (we recommend Anaconda distribution), Modern NVIDIA GPU, compute capability 5.0 of newer. CUDA 9.2, ASE( Atomic Simulation Environment)
GPUは、RTX 3090が搭載されたデスクトップパソコンがあれば、試験運用には、十分使えるのではないだろうか。
COMBO:東京大学が関係しているのだが、残念なのは少し古いことで、Python 2.7.xが使われている。
DeepChem:これは、オープンソースコードで、商用利用の制限も少なく、GitHubで管理されている。
DeepChem aims to provide a high quality open-source toolchain that democratizes the use of deep-learning in drug discovery, materials science, quantum chemistry, and biology.
DeepChem currently supports Python 3.7 through 3.8 and requires these packages on any condition. joblib, NumPy, pandas, scikit-learn, SciPy, TensorFlow, deepchem>=2.4.0 depends on TensorFlow v2, deepchem<2.4.0 depends on TensorFlow v1, Tensorflow Addons for Tensorflow v2 if you want to use advanced optimizers such as AdamW and Sparse Adam. (Optional)
チュートリアルも充実しているようである。
The DeepChem project maintains an extensive collection of tutorials. All tutorials are designed to be run on Google colab (or locally if you prefer). Tutorials are arranged in a suggested learning sequence which will take you from beginner to proficient at molecular machine learning and computational biology more broadly.
After working through the tutorials, you can also go through other examples. To apply deepchem to a new problem, try starting from one of the existing examples or tutorials and modifying it step by step to work with your new use-case. If you have questions or comments you can raise them on our gitter.
ある程度使ってみなければわからない。一見するとバイオ系のように思うが、筋道は同じようなものだろうと思うので、ある程度理解できるところまでは、チュートリアルに倣って足を踏み込んでみるのがよいかもしれない。
DeepChemのコードやツールはGitHubに置かれていて、GitHubは機械学習系の有用なコードやツールが膨大に蓄積されているので、適当に検索すれば、有用なツールやコードが容易に見つかる。重要なことは、目的を定めて、1つのサイトで良いから、チュートリアルや手順に従ってインストールし、自分で使えるところまで持っていくことである。ちょっとしたことで躓いて前に進まなくなることがあると思うが、簡単なコードで良いので、とにかく、jupyter notebook上でコードを走らせて結果を得るところまでやることが重要である。
9月4日(土)には、このDeepChemにチャレンジしてみよう。
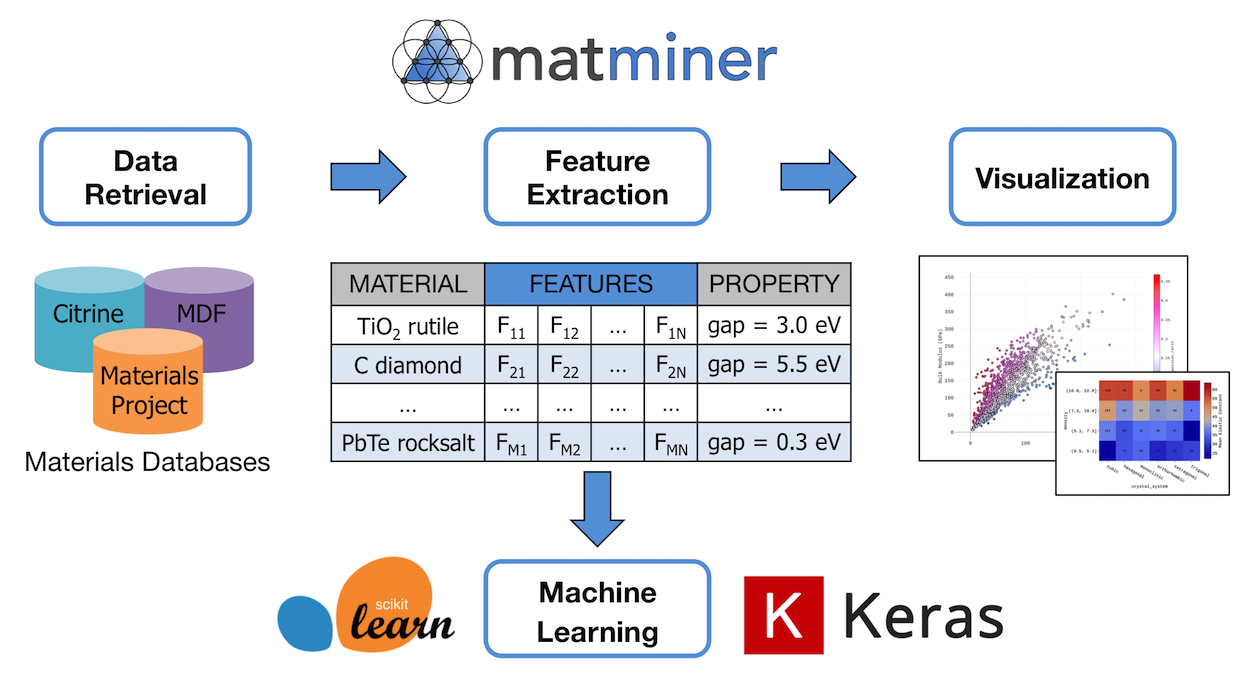
MatMiner:Table 12には、Python library for assisting machine learning in materials scienceと書かれている。MatMinerのホームページには、matminer is a Python library for data mining the properties of materials.と書かれていて、machine learningという言葉が含まれていない。下の方に次のように書かれている。
Matminer does not contain machine learning routines itself, but works with the pandas data format in order to make various downstream machine learning libraries and tools available to materials science applications.
MatMinerは、machine learningを含んでおらず、その出力がPandasのデータ形式なので、machine learningに容易に接続できるということのようである。次の図に、その関係が示されている。
9月4日(土)
DeepChemにチャレンジしよう!
チュートリアルが充実しているようだが、その紹介文を見ると、DeepChemはライフサイエンスの方を見ており、材料科学は付け足しのように見える。
このチュートリアルシリーズでは、DeepChemを使用して、ライフサイエンスの面白くてやりがいのある問題を解決する方法を学習します。このチュートリアルは、DeepChemの概要として、また分子機械学習、量子化学、バイオインフォマティクス、材料科学などのドメインにわたるさまざまな問題へのDeepChemの適用として機能します。このチュートリアルシリーズは、実装された新しいDeepChem機能とモデルで継続的に更新され、初心者がアクセスできるように設計されています。
次の説明では、創薬に最適なツールであると宣伝している。
1)キャリアアップ:ライフサイエンスにAIを適用することは、現在活況を呈している業界です。AIを中心とした大手製薬会社やバイオテクノロジー企業には、新たに資金提供を受けたスタートアップやイニシアチブが数多くあります。DeepChemを学び、習得することで、この分野の最前線に立ち、この分野でのキャリアに入る準備が整います。
2)人道上の考慮事項:病気は人間の苦しみの最も古い原因です。人類の文明の黎明期から、人類は病原体、癌、および神経学的状態に苦しんでいます。過去数世紀の最大の成果の1つは、多くの病気の効果的な治療法の開発でした。このチュートリアルのスキルを習得することで、過去の巨人の肩の上に立って新薬の開発を支援できるようになります。
3)薬のコストを下げる:新しい薬を開発する技術は、現在、専門家の小さなコアによってのみ実践できるエリートスキルです。創薬のためのオープンソースツールの成長を可能にすることで、これらのスキルを民主化し、創薬をより多くの競争に開放することができます。競争の激化は、薬のコストを下げるのに役立ちます。
チュートリアルには30以上の項目があって、大半はバイオ~タンパク質分子間相互作用~創薬だが、1つだけ、材料科学の項目がある。
Introduction To Material Science
One of the most exciting applications of machine learning in the recent time is it's application to material science domain. DeepChem helps in development and application of machine learning to solid-state systems. As a starting point of applying machine learning to material science domain, DeepChem provides material science datasets as part of the MoleculeNet suite of datasets, data featurizers and implementation of popular machine learning algorithms specific to material science domain. This tutorial serves as an introduction of using DeepChem for machine learning related tasks in material science domain.
最近の機械学習の最もエキサイティングなアプリケーションの1つは、材料科学分野へのアプリケーションです。 DeepChemは、機械学習の開発とソリッドステートシステムへの適用を支援します。機械学習を材料科学ドメインに適用する出発点として、DeepChemは、MoleculeNetデータセットスイートの一部として材料科学データセット、データ機能化ツール、および材料科学ドメインに固有の一般的な機械学習アルゴリズムの実装を提供します。このチュートリアルは、材料科学分野の機械学習関連タスクにDeepChemを使用する方法の概要として役立ちます。by Google翻訳
(MoleculeNet is a large scale benchmark for molecular machine learning. MoleculeNet curates multiple public datasets, establishes metrics for evaluation, and offers high quality open-source implementations of multiple previously proposed molecular featurization and learning algorithms (released as part of the DeepChem open source library). MoleculeNet benchmarks demonstrate that learnable representations are powerful tools for molecular machine learning and broadly offer the best performance.)
Traditionally, experimental research were used to find and characterize new materials. But traditional methods have high limitations by constraints of required resources and equipments. Material science is one of the booming areas where machine learning is making new in-roads. The discovery of new material properties holds key to lot of problems like climate change, development of new semi-conducting materials etc. DeepChem acts as a toolbox for using machine learning in material science.
伝統的に、実験的研究は、新しい材料を見つけて特徴づけるために使用されていました。しかし、従来の方法には、必要なリソースと機器の制約によって高い制限があります。材料科学は、機械学習が新たな道を切り開いている活況を呈している分野の1つです。新しい材料特性の発見は、気候変動、新しい半導体材料の開発など、多くの問題の鍵を握っています。DeepChemは、材料科学で機械学習を使用するためのツールボックスとして機能します。by Google翻訳
事例の1つは次の文献の内容である。
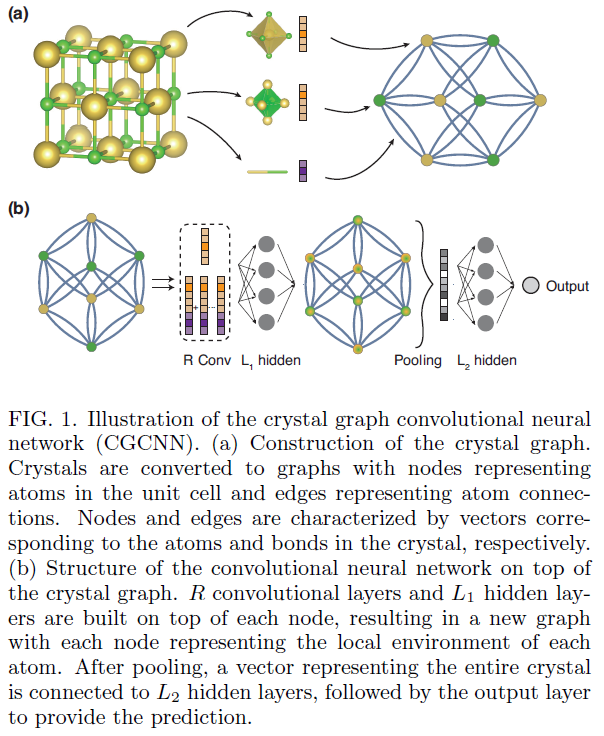
Crystal Graph Convolutional Neural Networks for an Accurate and Interpretable Prediction of Material Properties, Tian Xie and Je rey C. Grossman, arXiv:1710.10324v3 [cond-mat.mtrl-sci] 6 Apr 2018
Abstract :
The use of machine learning methods for accelerating the design of crystalline materials usually requires manually constructed feature vectors or complex transformation of atom coordinates to input the crystal structure, which either constrains the model to certain crystal types or makes it difficult to provide chemical insights. Here, we develop a crystal graph convolutional neural networks (CGCNN) framework to directly learn material properties from the connection of atoms in the crystal, providing a universal and interpretable representation of crystalline materials. Our method provides a highly accurate prediction of DFT calculated properties for 8 different properties of crystals with various structure types and compositions after trained with 10,000 data points. Further,
our framework is interpretable because one can extract the contributions from local chemical environments to global properties. Using an example of perovskites, we show how this information can be utilized to discover empirical rules for materials design.
Machine learning (ML) methods are becoming increasingly popular in accelerating the design of new materials by predicting material properties with accuracy close to ab-initio calculations, but with computational speeds orders of magnitude faster[1-3]. The arbitrary size of crystal systems poses a challenge as they need to be represented as a fixed length vector in order to be compatible with most ML algorithms. This problem is usually resolved by manually constructing fixed-length feature vectors using simple material properties[1, 3-6] or designing symmetry-invariant transformations of atom
coordinates[7-9]. However, the former requires case-by-case design for predicting different properties and the latter makes it hard to interpret the models as a result of the complex transformations.
In this letter, we present a generalized crystal graph convolutional neural networks (CGCNN) framework for representing periodic crystal systems that provides both
material property prediction with DFT accuracy and atomic level chemical insights.
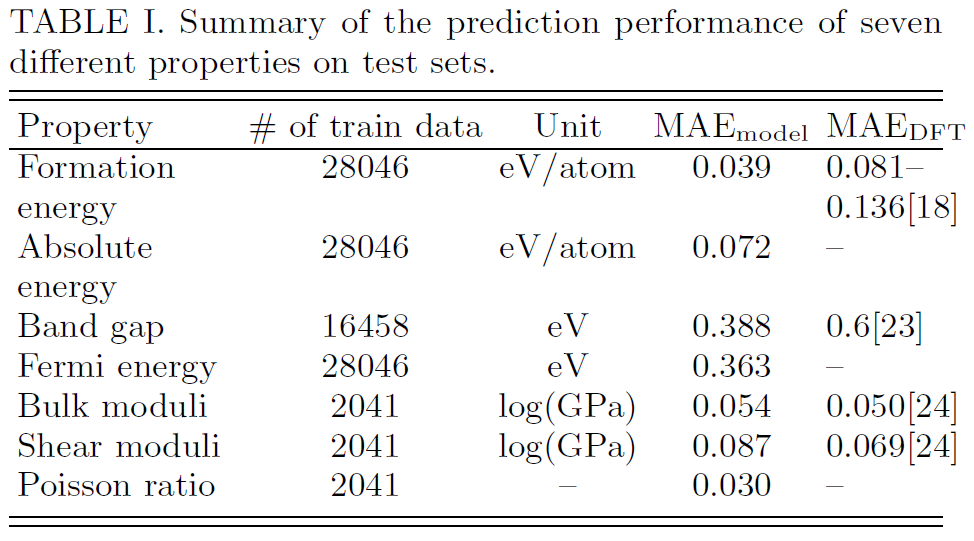
We summarize the performance in Table I and the corresponding 2D histograms in Figure
S4. As we can see, the MAE of our model are close to or higher than DFT accuracy relative to experiments for most properties when 10,000 training data is used.
In summary :
The crystal graph convolutional neural networks (CGCNN) presents a flexible machine learning framework for material property prediction and design knowledge extraction. The framework provides a reliable estimation of DFT calculations using around 10,000
training data for 8 properties of inorganic crystals with diverse structure types and compositions. As an example of knowledge extraction, we apply this approach to the design of new perovskite materials and show that information extracted from the model is consistent with common chemical insights and significantly reduces the search space for high throughput screening.
DeepChemの中に、CGCNNFeaturizerが含まれているので、importで呼び出して使うことができる。
9月7日(火)
CGCNNをもっとよく理解しよう。
DFT (density functional theory) : 密度汎関数理論と同程度の正確さで計算できる理由:
過去に、膨大な量の理論計算が進められてきたという背景がある。リチウムイオン電池の組成の最適化もその1例である。膨大な量の候補材料(候補になるかどうかを考えるよりも、できるだけ多くの種類の元素の組み合わせを計算機上で試してみることが重要であった)に対する徹底的な理論計算と特性値の計算結果を多数の研究者が計算結果を共有しながら進めてきたことによって、良い候補材料(単に性能が高いということだけでなく、信頼性、コスト、寿命などの種々の要因も並行して計算機上で理論計算をによって検討することも併せて行われることによって)が見つかった。
次の論文にそのことが正確に解説されている。
Commentary: The Materials Project: A materials genome approach to accelerating materials innovation, A. Jain et al., APL Materials 1, 011002 (2013)
Accelerating the discovery of advanced materials is essential for human welfare and sustainable, clean energy. In this paper, we introduce the Materials Project (www.materialsproject.org), a core program of the Materials Genome Initiative that uses high-throughput computing to uncover the properties of all known inorganic materials. This open dataset can be accessed through multiple channels for both interactive exploration and data mining. The Materials Project also seeks to create open-source platforms for developing robust, sophisticated materials analyses. Future efforts will enable users to perform ‘‘rapid-prototyping’’ of new materials in silico, and provide researchers with new avenues for cost-effective, data-driven materials design. © 2013 Author(s). All article content, except where otherwise noted, is licensed under a Creative Commons Attribution 3.0 Unported License. http://dx.doi.org/10.1063/1.4812323
I. INTRODUCTION
II. DATA GENERATION AND VALIDATION
III. DISSEMINATION: PROVIDING OPEN, MULTI-CHANNEL ACCESS TO MATERIALS INFORMATION
IV. ANALYSIS: OPEN-SOURCE LIBRARY
V. DESIGN: A VIRTUAL LABORATORY FOR NEW MATERIALS DISCOVERY
VI. CONCLUSION AND FUTURE
It is our belief that deployment of large-scale accurate information to the materials development community will significantly accelerate and enable the discovery of improved materials for our future clean energy systems, green building components, cutting-edge electronics, and improved societal health and welfare.
(deep learningがものすごい勢いで発展し始めたのが2012年であり、この解説が書かれた2013年の時点では、このmaterials genome approachがdeep learningによってさらに加速されるだろうということまでは予測されていなかったようである。2018年になって、materials genome approachによって蓄積されたDFT計算結果等は、CGCNNの学習のために活用され、次のレベルに進むことが可能になったということである。)
The SineCoulombMatrix featurizer a crystal by calculating sine coulomb matrix for the crystals. It can be called using dc.featurizers.SineCoulombMatrix function. [1]
The CGCNNFeaturizer calculates structure graph features of crystals. It can be called using dc.featurizers.CGCNNFeaturizer function. [2]
The LCNNFeaturizer calculates the 2-D Surface graph features in 6 different permutations. It can be used using the utility dc.feat.LCNNFeaturizer. [3]
SineCoulombMatrix featurizerとは何かを理解しておく必要がありそうなのでその論文を眺めてみよう。
Crystal Structure Representations for Machine Learning Models of Formation Energies, F. Faber et al., arXiv:1503.07406v1 [physics.chem-ph] 25 Mar 2015
We introduce and evaluate a set of feature vector representations of crystal structures for machine learning (ML) models of formation energies of solids. ML models of atomization energies of organic molecules have been successful using a Coulomb matrix representation of the molecule. We consider three ways to generalize such representations to periodic systems: (i) a matrix where each element is related to the Ewald sum of the electrostatic interaction between two different atoms in the unit cell
repeated over the lattice; (ii) an extended Coulomb-like matrix that takes into account a number of neighboring unit cells; and (iii) an ansatz that mimics the periodicity and the basic features of the elements in the Ewald sum matrix by using a sine function of the crystal coordinates of the atoms. The representations are compared for a Laplacian kernel with Manhattan norm, trained to reproduce formation energies using a data set of 3938 crystal structures obtained from the Materials Project. For training sets consisting of 3000 crystals, the generalization error in predicting formation energies of new structures corresponds to (i) 0.49, (ii) 0.64, and (iii) 0.37 eV/atom for the respective representations.
Materials Projectからのデータセットを用いて学習させているとのこと。
9月8日(水)
LCNNについての論文:
Lattice Convolutional Neural Network Modeling of Adsorbate Coverage Effects
Jonathan Lym et al., J. Phys. Chem. C 2019, 123, 31, 18951–18959
Abstract:
Coverage effects, known also as lateral interactions, are often important in surface processes, but their study via exhaustive density functional theory (DFT) is impractical because of the large configurational degrees of freedom. The cluster expansion (CE) is the most popular surrogate model accounting for coverage effects but suffers from slow convergence, its linear form, and its tendency to be biased toward the selection of smaller clusters. We develop a novel lattice convolutional neural network (LCNN) that improves upon some of CE’s limitations and exhibits better performance (test RMSE of 4.4 meV/site) compared to state-of-the-art methods, such as the CE assisted by a genetic algorithm and the convolution operation of the crystal graph convolutional neural network (CGCNN) (test RMSE of 5.5 and 6.8 meV/site, respectively) by 20–30%. Furthermore, LCNN can outperform other methods with less training data, implying accuracy with less DFT calculations. We analyze the van der Waals interaction via visualization of the hidden representation of the adsorbate lattice system in terms of individual site formation energies.
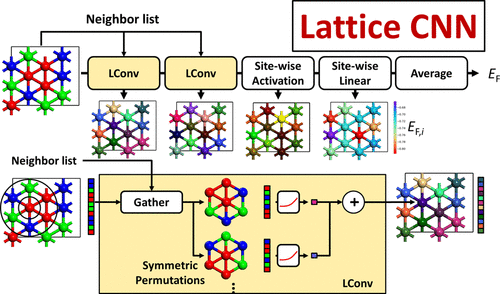
この論文は有料なので、残念だが、本文は読めないので、紹介できない。この図はAbstractに張り付けられているもので、説明はない。
著者らによる2ページ程度の要約版(講演要旨かもしれない)らしきものがあるのでそれを見てみよう。
Lattice Convolutional Neural Network for Modelling Adsorbate Coverage Effects
Jonathan Lym, Geun Ho Gu, Yousung Jung and Dionisios G. Vlachos
Introduction
Density Functional Theory (DFT) has revolutionized the field of catalysis by giving
researchers the ability to predict system properties at the quantum level at reasonable accuracy and computational cost. However, DFT still has its limitations and performs poorly for some systems, such as studying coverage effects due to the large size of systems and the vast configurational degrees of freedom. To overcome these limitations, surrogate models are trained using DFT calculations to reduce the computational cost further without significantly sacrificing accuracy. The most popular model to study coverage effects is the cluster expansion (CE), which is a linear lattice-based model that models long and short-range interactions. While it has been used widely in the literature, the CE suffers from slow convergence due to adsorbates moving from ideal lattice positions, lateral interactions having nonlinear forms, and the CE’s heuristics’ tendency to prefer small clusters with short-range interactions that may not be sufficient to fully capture the local environment.
In this work, we develop a novel lattice graph convolutional neural network (LGCNN) and
compare it to the cluster expansion trained using three different cluster selection techniques (heuristics, the least absolute shrinkage and selection operator (LASSO), and the genetic algorithm (GA)) and the crystal graph convolutional neural network (CGCNN) implemented by Xie and Grossman for a multi-adsorbate system (O and NO on Pt(111)).
Materials and Methods
The configurations and DFT data used to train, validate, and test the machine learning
models of the system were provided by Bajpai et al. The configurations were reoptimized with the Vienna Ab initio Simulation Package (VASP) using the PBE+D3 functional to observe the effect of van der Waals forces on formation energies. The heuristic and LASSO regression models were implemented with in-house Python code using the Scikit-learn library. The Alloy-Theoretic Automated Tookit (ATAT) was used as the GA model.
The CGCNN and the LGCNN models were created using Tensorflow. To evaluate each
model, 10% of the data was withheld for testing. The remaining 90% was used to optimize hyperparameters and train the models using 10-fold cross validation.
Results and Discussion
Figure 1 shows the training and test error of each method as a function of the fraction of data used for training. When all the training data is used, the LGCNN has a test root mean squared error (RMSE) of 2.14 meV/site and outperforms the other methods. The
LGCNN has a lower test RMSE than the other methods when using only 40% of the training data. This superior performance is attributed to the nonlinear convolution operator learning the local environment around each site effectively.
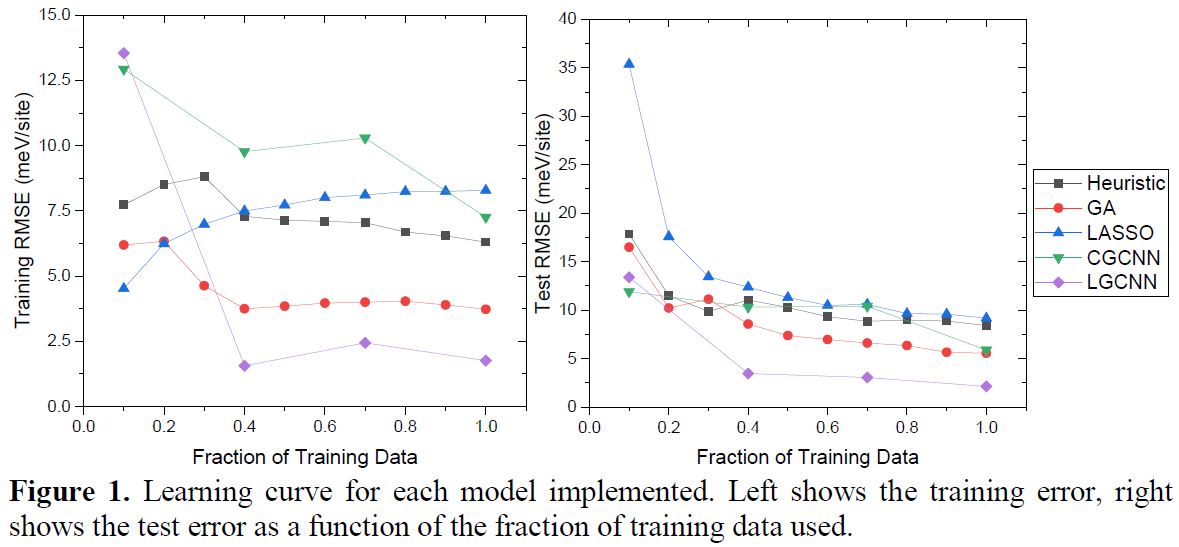
吸着状態の構造最適化の重要性を示しているのだろうと思う。反応中間体のエネルギー状態が、より現実に近い値として計算できていると推測される。
The configurations and DFT data used to train, validate, and test the machine learning
models of the system were provided by Bajpai et al.
吸着分子の吸着配置構造データとDFTデータはBaipaiらによって提供しているとのことなので、引用されているBaipaiらの論文をチェックする。有料なのでアブストラクト(図面付き)だけだが、みてみよう。
Binary Approach to Ternary Cluster Expansions: NO–O–Vacancy System on Pt(111)
A. Bajpai, K. Frey and W. F. Schneider, J. Phys. Chem. C 121, 13, 7344 (2017)
Abstract
Cluster expansions (CEs) provide an exact framework for representing the configurational energy of interacting adsorbates at a surface. Coupled with Monte Carlo methods, they can be used to predict both equilibrium and dynamic processes at surfaces. In this work, we propose a three-binary-to-single-ternary (TBST) fitting procedure, in which a ternary CE is approximated as a linear combination of the three binary CEs (O–vac, NO–vac, and NO–O) obtained by fitting to the three binary legs. We first construct a full ternary CE by fitting to a database of density functional theory (DFT) computed energies of configurations across a full range of adsorbate configurations and then construct a second ternary using the TBST approach. We compare two approaches for the NO–O–vacancy system on the (111) surface of Pt, a system of relevance to the catalytic oxidation of NO. We find that the TBST model matches the ternary CE to within 0.018 eV/site across a wide range of configurations. Further, surface coverages and NO oxidation rates extracted from Monte Carlo simulations show that the two models are qualitatively consistent over the range of conditions of practical interest.
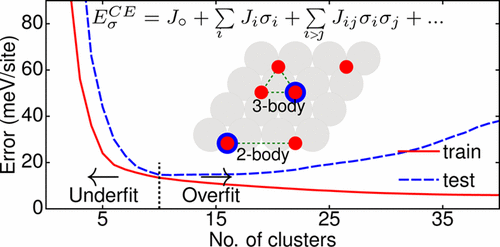
同一の系について計算しているようなので、J. Lymらは、新たに開発したLGCNNを用いることによって(それだけではないようだが)誤差を1/10くらいにまで減少させることができたということのようである。
LCNNと同様に、吸着(不均一系触媒表面における物理化学現象)を扱っている(ACE-GCN)というのが2021年に発表されたようである。
Adsorbate chemical environment-based machine learning framework for heterogeneous catalysis, P. G. Ghanekar et al., 10.33774/chemrxiv-2021-8fcxm
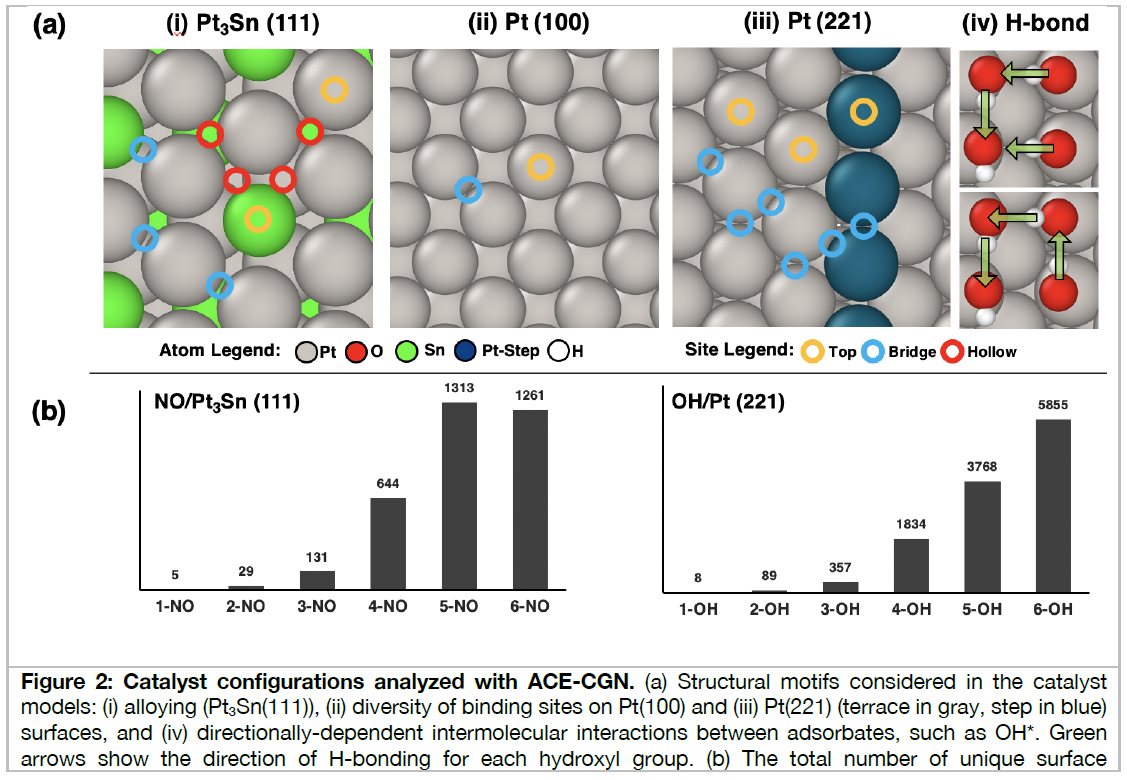
Heterogeneous catalytic reactions are influenced by a subtle interplay of atomic-scale factors, ranging from the catalysts’ local morphology to the presence of high adsorbate coverages. Describing such phenomena via computational models requires generation and analysis of a large space of surface atomic configurations. To address this challenge, we present the Adsorbate Chemical Environment-based Graph Convolution Neural Network (ACE-GCN), a screening workflow that can account for atomistic configurations comprising diverse adsorbates, binding locations, coordination environments, and substrate morphologies. Using this workflow, we develop catalyst surface models for two illustrative systems: (i) NO adsorbed on a Pt3Sn(111) alloy surface, of interest for nitrate electroreduction processes, where high adsorbate coverages combine with the low symmetry of the alloy substrate to produce a large configurational space, and (ii) OH* adsorbed on a stepped Pt(221) facet, of relevance to the Oxygen Reduction Reaction, wherein the presence of irregular crystal surfaces, high adsorbate coverages, and directionally-dependent adsorbate-adsorbate interactions result in the configurational complexity. In both cases, the ACE-GCN model, having trained on a fraction (~10%) of the total DFT-relaxed configurations, successfully ranks the relative stabilities of unrelaxed atomic configurations sampled from a large configurational space. This approach is expected to accelerate development of rigorous descriptions of catalyst surfaces under in-situ conditions.
2番目の事例は、白金触媒表面における酸素の還元反応、すなわち、燃料電池のカソード電極における酸素還元反応(ORR)の原子・分子レベルでの反応機能の解明のためのDFT・機械学習モデルの研究開発成果であり、高性能触媒開発につながるものである。ACE-GCNモデルを使いこなせるようになるために学ぼう。
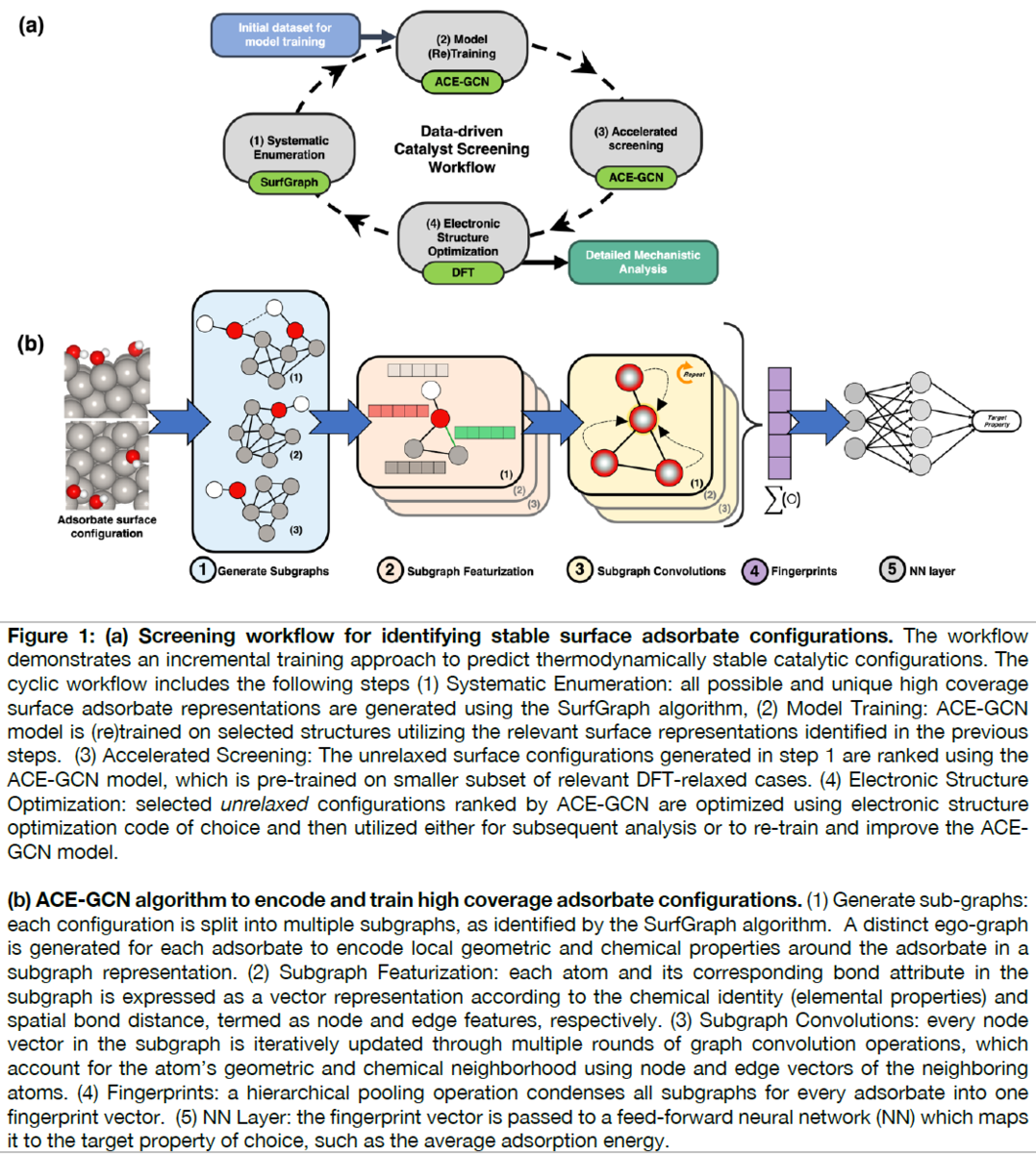
Workflow and ACE-GCN Framework (Adsorbate Chemical Environment-based Graph Convolution Neural Network)
Figure 1(A) summarizes the proposed screening framework. The cyclic workflow is divided into four parts:
(i) systematic enumeration of unique atomic configurations,
(ii) (re)training the surrogate model with data of incremental complexity,
(iii) accelerated screening using the surrogate model to identify the most relevant configurations amongst possible geometries, and
(iv) electronic structure relaxation of selected structures, which can be used for in-depth mechanistic analysis, or to improve the surrogate model.
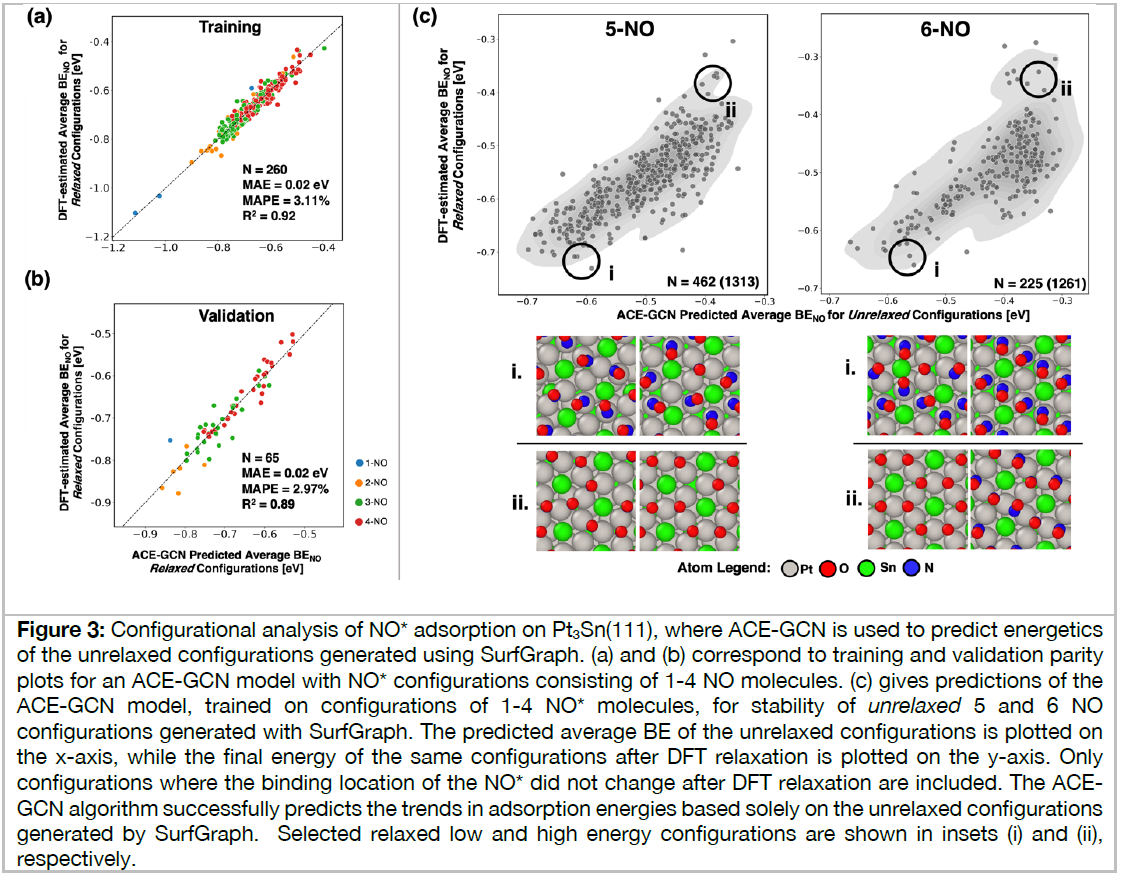
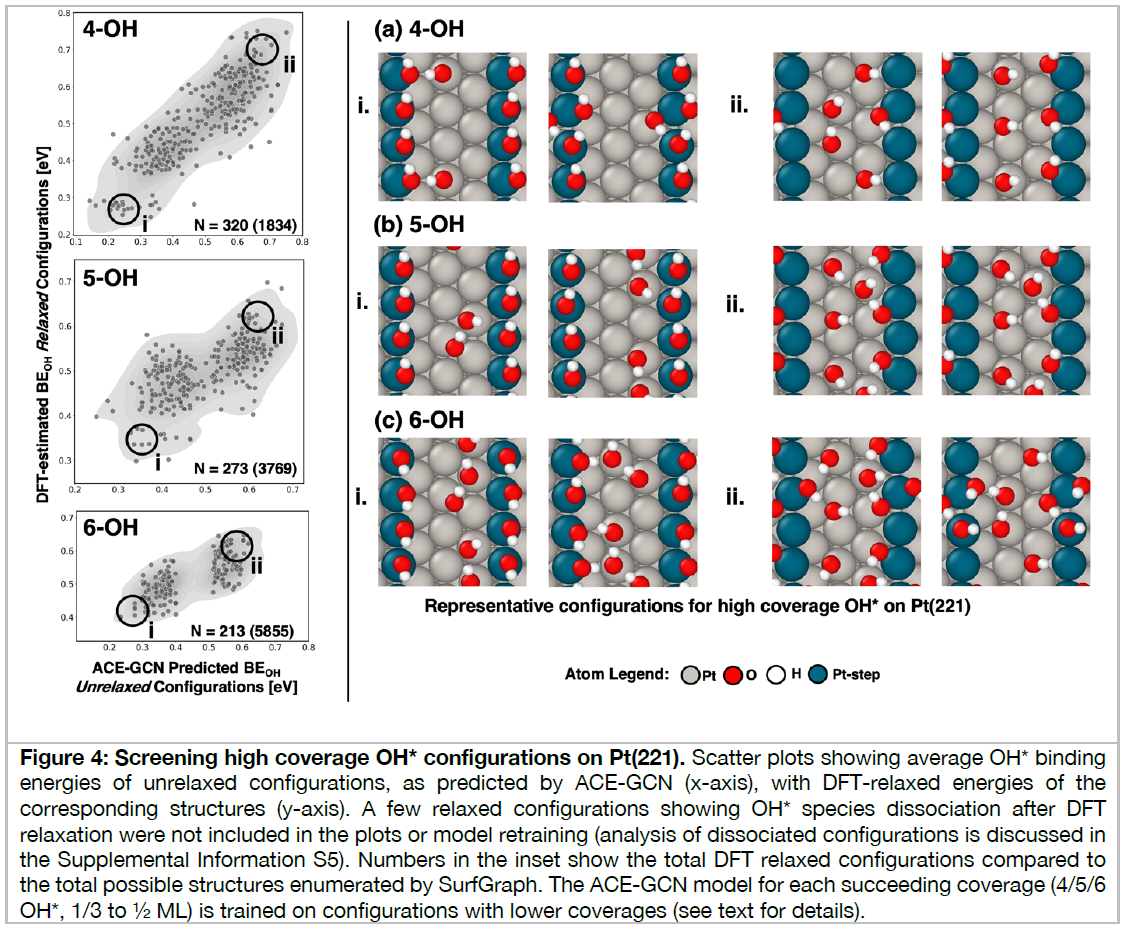
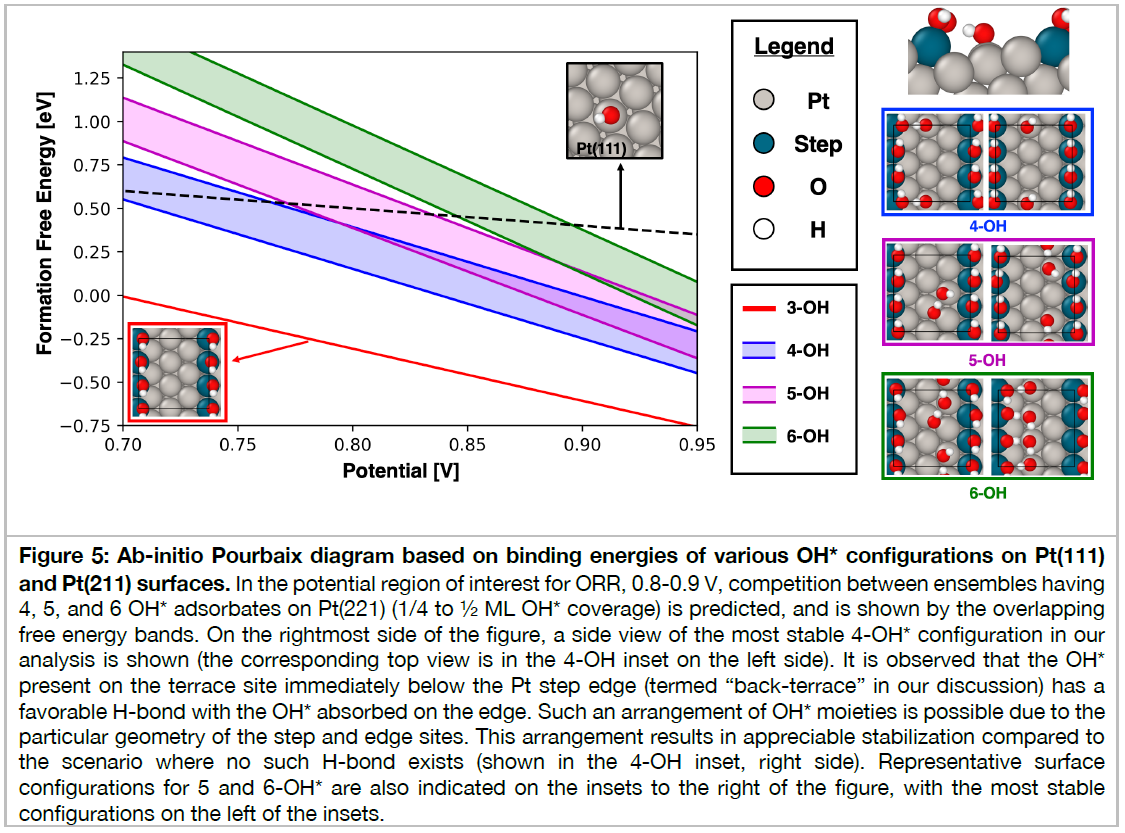
約600行の文章と5枚の図面および55件の文献を今月中に理解しよう。
Introduction 38行~51行:
不均一系触媒の複雑な分子レベルの詳細を解明するためには、理論的な計算モデルが、不可欠になってきている。スケーリングやブレンステッド-エヴァン-ポランニー (Brønsted-Evan-Polanyi) 関係などの記述子ベースの相関関係と組み合わせたハイスループット材料スクリーニング戦略[1–4]は、重要な酸素、窒素、および炭素ベースの化学物質の有望な候補を特定する上で中心的な役割を果たしてきた。
1. Greeley, J. et al. Alloys of platinum and early transition metals as oxygen reduction electrocatalysts. Nature Chemistry 1, 552–556 (2009).
2. Bligaard, T. et al. The Brønsted–Evans–Polanyi relation and the volcano curve in heterogeneous catalysis. Journal of Catalysis 224, 206–217 (2004).
3. Nørskov, J. K. et al. Origin of the Overpotential for Oxygen Reduction at a Fuel-Cell Cathode. The Journal of Physical Chemistry B 108, 17886–17892 (2004).
4. Lansford, J. L., Mironenko, A. V. & Vlachos, D. G. Scaling relationships and theory for vibrational frequencies of adsorbates on transition metal surfaces. Nature Communications 8, 016105 (2017).
122行~
First, adsorbate configurations are generated by enumerating adsorbate binding locations on the catalyst surface using the SurfGraph algorithm.
This algorithm utilizes graph-based representations to identify and create unique surface adsorbate configurations, systematically accelerating the task of generating complex catalytic model motifs.
触媒表面において吸着物質がどのように配置しているかを決める必要がある。触媒表面には面方位によって固有の原子配列があり、吸着物質が触媒表面の原子配列のどこにどのように配置するのかを決める。吸着物質は複数存在しそれらの配置の仕方も決めなければならない。研究者の経験や勘、直観だけでは可能性のある配置を網羅することは不可能であり、その代わりをするアルゴリズムとしてSurfGraphがある。
23. Deshpande, S., Maxson, T. & Greeley, J. Graph theory approach to determine configurations of multidentate and high coverage adsorbates for heterogeneous catalysis. npj Computational Materials 6, 79 (2020).
24. Boes, J. R., Mamun, O., Winther, K. & Bligaard, T. Graph Theory Approach to High-Throughput Surface Adsorption Structure Generation. The Journal of Physical Chemistry A 123, 2281–2285 (2019).
***中断***
